что это такое и зачем НКО их писать
Что такое лонгриды, как с их помощью можно рассказать историю, и для чего НКО нужен этот формат?
Поделитесь этой статьей с друзьями
Что такое лонгрид
Лонгриды (от англ. «long read» – длинное чтение) – достаточно новый формат, появившийся в медиа несколько лет назад и изменивший наши представления о чтении текстов в Интернете.
Лонгриды, длинные, глубокие истории, лучше всего читать вдалеке от рабочего стола – по дороге на работу, в самолете или на диване – тогда, когда у вас есть время для того, чтобы полностью погрузиться в чтение.
Лонгриды – это идеальный формат для iPad, iPhone или Kindle и таких приложений, как Read It Later, Flipboard и Instapaper, с помощью которых вы можете сохранить интересные вам тексты и прочитать их в любое удобное время.
Журналистские статьи, короткие рассказы, интервью, исторические документы – все это может быть лонгридом (как правило, длина лонгрида больше 1 500 слов).
Какими бывают лонгриды
Классическим примером лонгрида считается проект New York Times «Snowfall». Интерактивная история про лыжников, которые застряли в ловушке под лавиной в горах в Вашингтоне, стала одним из лучших медийных проектов 2012 года и на долгое время определила формат лонгридов.
Первая часть лонгрида «Snowfall». Изображение: nytimes.comЗа «Snowfall» последовали и другие лонгриды, привлекшие тысячи просмотров и ставшие образцами жанра.
Так, газета The Guardian выпустила интерактивный материал «NSA Files: Decoded», освещающий разглашение секретных документов, сделанное Эдвардом Сноуденом; в материале National Geographic «Killing Kennedy» разворачиваются две истории – история становления Джона Кеннеди президентом и история разочарования бывшего морского пехотинца Освальда в США; New York Time опубликовал интерактивное путешествие из Петербурга в Москву – «The Russia Left Behind»; «Медуза» к годовщине операции по присоединению Крыма выпустила спецпроект «Вежливые люди», в котором изучила феномен «вежливых людей» со всех сторон.
Лонгриды не обязательно посвящены острым политическим и социальным темам, но, как правило, именно они вызывают наибольший общественный резонанс.
Для чего НКО нужно писать лонгриды
1. Возможность рассказать историю
Лонгрид – это, прежде всего, возможность рассказать вашу историю. Истории помогают нам общаться друг с другом, убеждать и вдохновлять. Истории запоминаются нам лучше, чем что-либо, потому что затрагивают наши чувства и эмоции, и именно историями мы чаще всего делимся друг с другом.
Маршалл Ганц, участник многих политических кампаний и активист социальных движений, считает, что истории – это сердце социальных изменений:
У всех социальных движений есть истории, и они очень важны. Участвовать в социальном движении часто значит рисковать, быть неуверенным, идти против большинства. Откуда же брать смелость? Где искать надежду?
Все это можно найти в нарративах, в тех историях, которые рассказывают о жизни людей, их работе, вере, традициях, а лонгриды – один из инструментов, с помощью которых мы можем донести эту важную информацию до большого числа людей.

2. Возможность рассказать историю полностью
Благодаря своей длине лонгриды позволяют рассказать историю полностью, со всеми важными деталями и значимыми подробностями.
Так, лонгрид о новой теории аутизма насчитывает более 7 000 слов не потому, что кто-то смог написать 7 000 слов вместо 700, а потому, что именно столько было необходимо, чтобы хорошо рассказать об этом.
С помощью лонгрида вы можете затрагивать темы, о которых нельзя рассказать коротко, – большие и сложные идеи, истории людей, удивительные и малоизвестные вещи и концепции.
3. Возможность использовать различные мультимедиа
Безусловным преимуществом лонгрида является возможность использовать различные мультимедиа – вы можете добавить к вашему тексту большие качественные фотографии, занимающие весь экран, интерактивную инфографику, фоновые звуки, видеоинтервью с участниками истории.
Все это не только дополнит вашу историю, но и создаст у читателей чувство вовлеченности и погружения в материал и позволит удерживать их внимание на протяжении всего повествования.
4. Возможность привлечь к истории больше внимания
Еще одним преимуществом лонгридов является более высокий рейтинг в поисковых запросах и большая, по сравнению с обычными материалами, популярность в социальных сетях.
Согласно исследованию первые 10 результатов поисковых запросов, как правило, являются материалами длиннее 2 000 слов, а лонгриды, опубликованные в социальных сетях, получают в среднем больше лайков и репостов, чем небольшие материалы.
Это значит, что больше людей смогут увидеть и прочитать ваш лонгрид.
5. Возможность создать что-то значимое
В эпоху быстрого просмотра статей, большая часть из которых почти также быстро забывается и теряется среди миллионов подобных текстов в Интернете, хорошие лонгриды имеют больше шансов остаться надолго и стать материалами, к которым будут возвращаться и на которые будут ссылаться.
Такие популярные ресурсы, агрегирующие лонгриды, как Longreads, девайсы и приложения для чтения, с помощью которых можно сохранить текст и всегда иметь к нему доступ, делают жизненный цикл лонгридов значительно длиннее обычных материалов и заметок.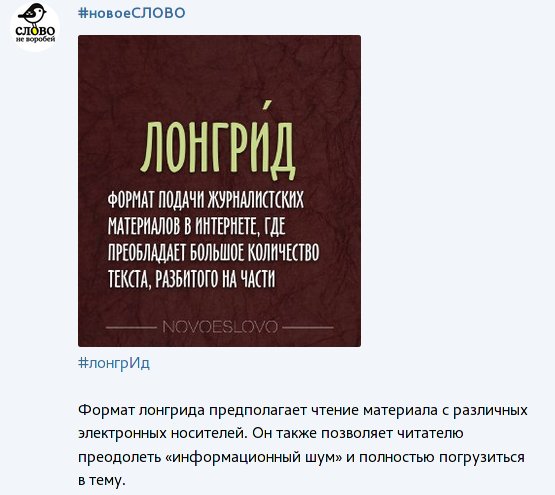
Создание качественного лонгрида – это тяжелая работа, требующая много времени, но, в конечном итоге, затраченные усилия окупаются тем распространением и эффектом, которого удалось достичь.
Где опубликовать лонгрид
Вы можете опубликовать лонгрид в вашем блоге и на сайте или использовать для этого такие специальные платформы, как Medium, Readymag и Stampsy.
Лонгрид — что это и как его создать. Формат и примеры длинных статей
Считается, что длинные тексты – не для современного мира. Существует расхожее мнение, что из-за синдрома дефицита внимания, клипового мышления, а также привычки потреблять максимально простой развлекательный контент, люди разучились усваивать что-то большее, чем пост ВКонтакте. Это действительно похоже на правду, учитывая, что мы гораздо чаще скролим ленту, нежели читаем книги. И, тем не менее, примитивные онлайн-статейки скоро окончательно канут в Лету.
На смену им приходит лонгрид – объёмный информативный материалы на 10 000 символов и больше. Кажется, что еще немного – и лонгриды по популярности обгонят интернет-мемы. Давайте разбираться, в чем причина этого парадокса.
Кажется, что еще немного – и лонгриды по популярности обгонят интернет-мемы. Давайте разбираться, в чем причина этого парадокса.
Рекомендуем: Click.ru – маркетплейс рекламных платформ:
- Более 2000 рекламных агентств и фрилансеров уже работают с сервисом.
- Подключиться можно самому за 1 день.
- Зарабатывайте с первого потраченного рубля, без начальных ограничений, без входного барьера.
- Выплаты на WebMoney, на карту физическому лицу, реинвестирование в рекламу.
- У вас остаются прямые доступы в рекламные кабинеты, рай для бухгалтерии по документообороту и оплатам.
Читайте также: 10 лучших курсов для начинающих копирайтеров
Что такое лонгрид?
Ответ на этот вопрос кроется в самом слове «лонгрид». С английского long read переводится как «долгое чтение».
В Сети реально найти лонгрид на любую тему и в любом стиле. Поэтому, чисто гипотетически, существуют сотни разновидностей лонгридов. Рассмотрим наиболее ходовые.
Лонгрид-портрет
Используется для презентации звезд шоу-бизнеса, коучей, бизнесменов, преподавателей известных школ и других публичных персон. Здесь автор должен учитывать, что люди обожают читать чужие биографии и истории успеха. Если крутой фотограф расскажет, как он выживал на копейки в студенческой общаге, собирал деньги на первую камеру, получал недоуменные взгляды от родственников, когда решил бросить инженерно-технический факультет, и с боем отвоевывал первых заказчиков, статья про него разойдется просто потому, что она вдохновляет и мотивирует. Среди тысяч замотивированных наверняка найдется пара десятков новых клиентов и пара сотен тех, кто запомнит фотографа и посоветует его своим друзьям. В данном случае, лонгрид – работа на имя.
Лонгрид-репортаж
Пожалуй, эта разновидность лонгридов наиболее близка к старой-доброй журналистике. Репортажем называют подробный обзор какого-либо события через некоторое время после того, как оно произошло. За примерами далеко ходить не надо. На любом новостном сайте, вроде «Медузы» или The Village вы найдете десятки подобных статей.
Лонгрид-реконструктор
Писать можно не только о том, что случилось недавно. Реконструктор – это текст, посвященный событию, явлению, персонажу или произведению из прошлого. Здесь дается наибольший простор для творчества. Можно хоть добавлять аудиодорожки, хоть цитировать стихи перед каждым подразделом, хоть оформлять повествование в духе художественного рассказа или криминальной хроники – так будет только интереснее.
Реконструктор – это текст, посвященный событию, явлению, персонажу или произведению из прошлого. Здесь дается наибольший простор для творчества. Можно хоть добавлять аудиодорожки, хоть цитировать стихи перед каждым подразделом, хоть оформлять повествование в духе художественного рассказа или криминальной хроники – так будет только интереснее.
Коммерческий лонгрид
Видели огромные описания товаров с фото, инфографикой и видеообзорами? Это – новая эффективная форма продаж. Коммерческий лонгрид на сайте способен повысить доверие и к продукции, и к продавцу. Представьте, что вы ищете в Интернете новый смартфон. На какую модель вы скорее обратите внимание – на ту, которой посвящен детальный разбор с тестировкой, снимками со всех сторон и отзывами довольных покупателей, или на ту, которая описана одним сухим абзацем?
Мультимедийный лонгрид
Это – статьи, щедро разбавленные картинками, анимацией, яркими надписями, видеороликами и т. д. Собственно, любой из лонгридов можно отчасти отнести к этому типу, однако здесь есть один важный нюанс. Как правило, в лонгридах главную роль играет текст – остальной «декор» его только дополняет. В мультимедийной же версии все детали равноправны, а иногда текст идет связующим звеном между графическими элементами.
Абсолютно все типы лонгридов способны приносить прибыль владельцам. Здорово написанная длинная статья умеет продавать не хуже landing page. Как минимум, ее преимущество в том, что она не устаревает и может работать годами. И это не говоря о вирусном эффекте, повышении доверия к сайту и увеличении посещаемости.
Почему формат лонгрида так популярен?
Мы уже поняли, чем хороши лонгриды для владельцев сайтов, но почему их так любят читатели? Чтобы выяснить подлинную причину популярности лонгридов, нужно провести масштабное социологическое исследование с выборкой, опросами и анализом данных. Пока мы можем строить лишь более-менее правдоподобные теории.
Во-первых, лонгриды отчасти выступают заменителями книг. Существует немалая часть пользователей Сети, которые осознают ценность чтения и пытаются заниматься саморазвитием, но им некогда разбирать полноценные монографии.
Во-вторых, этот формат текста рассчитан на поэтапное ознакомление в перерывах между важными делами. Лонгриды прекрасно читаются людьми, которые стоят в очередях, сидят на скучных парах, едут в транспорте, прокрастинируют во время работы. А если нет времени доскроллить прямо сейчас, лонгрид всегда можно репостнуть себе на страницу и вернуться к нему вечером, а то и через месяц.
В-третьих, читать лонгрид по любимой теме попросту увлекательно. Мастерски написанный текст, интригующие подзаголовки, подходящие иллюстрации – и 45 минут пролетают незаметно.
При подготовке лонгридов важно учитывать эти особенности. Ориентируйтесь на то, что вызовет отклик у вашей целевой аудитории, пишите так, как будто создаете художественное произведение. Если нужно, потратьте на это месяц и больше – главное, чтобы в итоге ваша статья выстрелила, а не стала одной из миллиона.
По какой структуре создавать лонгриды?
Опытные онлайн-журналисты настаивают на том, что лонгриды нельзя делать по лекалам. По их мнению, лонгрид – это авторский материал, поэтому здесь нет места жестким правилам, как в написании продающих текстов. Это действительно так, и смелые решения только приветствуются, но, тем не менее, базовую структуру никто не отменял. Когда приступите к своему первому лонгриду, учитывайте следующие элементы.
Время чтения
Маленькая, но важная деталь, про которую многие забывают. А зря. Строчка, где указывается, сколько минут уйдет на статью, не только выражает заботу о публике, но и увеличивает количество дочитываний. Человек сразу прикидывает, сможет ли он выделить на лонгрид полчаса или больше, и, ориентируясь на это, спокойно погружается в текст.
Чтобы узнать сроки, включите таймер, откройте готовый материал и перечитайте его в обычном темпе. Не обязательно фанатично считать секунды – достаточно указать приблизительную цифру.
Интригующее название
Забудьте на время о продающих заголовках. В лонгридах не нужны выгоды, преимущества и цифры. Здесь главное – запоминаемость, нетривиальность и отражение смысла статьи. Это – хороший шанс продемонстрировать свою начитанность и творческие способности, однако старайтесь избегать штампов и трюизмов. Если вам в голову пришла строчка, которую вы уже сто раз видели на первой полосе, лучше замените ее чем-нибудь другим.
Оглавление статьи
Перед объемной статьей крайне желательно указать гиперссылки на каждый пункт. Помните, что значительная часть публики не сможет прочитать лонгрид полностью. Дать людям возможность выхватить нужную информацию – гораздо лучше, чем ничего.
Вступление
Укажите читателю, о чем будет идти речь в тексте, и плавно подведите его к основной части. Чтобы красиво начать лонгрид, можно использовать:
- цитаты из известных произведений;
- шутки;
- пословицы и поговорки;
- метафоры и аллегории
Вступление должно быть максимально плотным.
Основная часть
«Тело» больших текстов обязательно включает в себя несколько разделов. Особых требований к объемам нет, однако старайтесь, чтобы каждый раздел был коротким (примерно 2 тысячи символов). Людям психологически легче усваивать небольшие порции информации. Если у вас получился объемный параграф, где важна каждая строка, еще раз подумайте, как его можно препарировать без ущерба для смысла.
Каждая часть начинается с подзаголовка. Делайте их так же, как и название статьи – передавайте суть и используйте нетривиальный подход.
Вывод
Не думайте, что ваша аудитория не дочитает лонгрид до конца, поэтому на выводы можно забить. Это – распространенная ошибка многих авторов, которые испортили не один десяток текстов, прежде чем поняли, что было не так. Беспомощный финал гарантированно погубит впечатление даже от самой лучшей статьи.
Беспомощный финал гарантированно погубит впечатление даже от самой лучшей статьи.
Чтобы сделать последний абзац сильным, перечитайте готовый материал несколько раз и подумайте над тем, как его резюмировать. Опять же, пытайтесь выйти за рамки банальности. Необычная мысль, возникшая в конце, гораздо лучше скучного пересказа вышеизложенного.
Призыв к действию
Call to action в лонгридах – это просьба поставить лайк и сделать репост в соцсетях. Часто его публикуют в конце, но он будет уместен и вначале, и в середине.
Смотрите по теме: Призыв к действию в рекламе
Мультимедийные файлы
Дополнительные материалы привлекают внимание и помогают читателям легче доскроллить материал до конца. Поэтому сделайте так, чтобы:
- картинки, фото, гифки и все остальные мелочи были распределены по повествованию равномерно;
- они были в тему.
Это интересно: Грамотная структура текста
Продвижение лонгрида
Как вы уже поняли, лонгрид можно создавать чуть ли не под любые цели. Лонгриды используют для рекламы себя как специалиста, для презентации товара, для заполнения научных ресурсов, для освещения актуальных событий и т. д. Однако сама по себе статья не будет работать в полную силу. Чтобы привлечь внимание большего количества народа, ее необходимо грамотно продвинуть. Рассмотрим несколько бесплатных способов.
Лонгриды используют для рекламы себя как специалиста, для презентации товара, для заполнения научных ресурсов, для освещения актуальных событий и т. д. Однако сама по себе статья не будет работать в полную силу. Чтобы привлечь внимание большего количества народа, ее необходимо грамотно продвинуть. Рассмотрим несколько бесплатных способов.
- Добавление SEO-ключей. Google и Яндексу нравятся длинные тексты. Если вы грамотно подберете семантическое ядро, есть хорошие шансы попасть на первое место поисковой выдачи.
- Распространение в соцсетях. То самое «ставьте лайк и делайте репост», о котором мы уже говорили. Чтобы облегчить читателям задачу, разместите рядом с призывом линки на ВКонтакте, Facebook и другие соцсети.
- Тематические сообщества. Админам пабликов нужно ежедневно поставлять контент аудитории. Они с удовольствием публикуют чужие статьи, если они действительно полезные и интересные.
- Публикация на Яндекс.
 Дзен. Регистрация канала на Яндекс.Дзене и перенаправление туда материалов со своего сайта – отличный способ поднять трафик и получить сотни, а то и тысячи новых постоянных читателей. Правда, там есть свои подводные камни в виде беспричинных банов и пессимизации аккаунта, но в любом случае вы ничего не потеряете.
Дзен. Регистрация канала на Яндекс.Дзене и перенаправление туда материалов со своего сайта – отличный способ поднять трафик и получить сотни, а то и тысячи новых постоянных читателей. Правда, там есть свои подводные камни в виде беспричинных банов и пессимизации аккаунта, но в любом случае вы ничего не потеряете.
Как видите, лонгриды раскручивают сами себя. Если текст действительно хороший, он не останется незамеченным в Сети.
Заключение
Итак, чтобы создать лонгрид, который захочется прочитать до конца, нужно три вещи: экспертность, хороший слог и личность автора. Досконально изучайте предмет беседы, пишите только о том, что вам близко, делайте так, чтобы ваши слова приносили пользу людям. Читайте художественную литературу, писательские блоги и чужие лонгриды, практикуйтесь в письме – в идеале, пишите не менее тысячи слов каждый день, как советовал Рэй Бредбери. Развивайте свой вкус и стиль, учитесь мыслить нестандартно, высказывайте смелые идеи.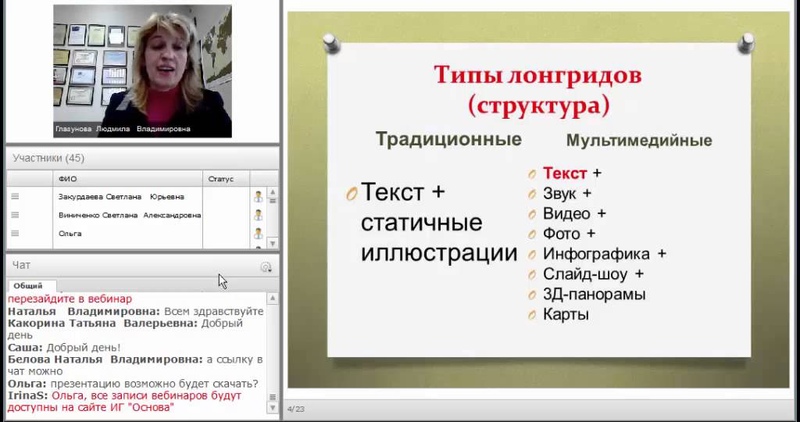 Пару лет в таком режиме – и вы сможете не только зарабатывать большие деньги на текстах, но и станете уважаемым автором, за которого будут драться редакции топовых журналов. Оно же того стоит?
Пару лет в таком режиме – и вы сможете не только зарабатывать большие деньги на текстах, но и станете уважаемым автором, за которого будут драться редакции топовых журналов. Оно же того стоит?
Лонгрид — что это такое, как написать его интересно, структура и примеры
Поделиться статьёй:
Принято считать, что объемные статьи длиной более десяти тысяч знаков никогда не будут пользоваться большой популярностью — пользователи интернета и социальных сетей привыкли к небольшим информативным текстам, на которые не придется тратить свое драгоценное время. Но в последнее время разрушить этот стереотип удается лонгридам: длинные тексты, написанные особым образом, действительно начали привлекать внимание большой аудитории — к такому заключению пришли исследователи. Именно поэтому стоит уделить особое внимание особенностям написания таких материалов.
Лонгрид: что это?Лонгриды создаются для действительно длительного чтения. Но просто написать статью со множеством слов — это не значит написать Longread. Важно суметь рассказать интересную историю, в которую читатель сможет окунуться с головой, прочитав статью до конца. Обычно объем таких текстов начинается от 1000-1200 слов: известно, что самые популярные лонгриды достигают размера в 3000 слов!
Но просто написать статью со множеством слов — это не значит написать Longread. Важно суметь рассказать интересную историю, в которую читатель сможет окунуться с головой, прочитав статью до конца. Обычно объем таких текстов начинается от 1000-1200 слов: известно, что самые популярные лонгриды достигают размера в 3000 слов!
Для удобства работы с такими текстами на любых носителях авторы оформляют их особым способом: делят на смысловые части и добавляют мультимедийные вставки, среди которых могут быть видео- и фотоматериалы, опросы и инфографика, а также музыка и многое другое — такой подход к написанию лонгрида помогает сделать его легким для чтения и восприятия.
Longread и его преимуществаНесмотря на то, что такие тексты не являются рекламными и не содержат в себе продающих фраз, с их помощью можно превратить обычного пользователя страницы в покупателя: главное, чтобы текст был качественным. Итак, лонгриды имеют ряд преимуществ и особенностей:
- они наиболее качественно концентрируют на себе внимание пользователей;
- такой формат легко воспринимается любителями выходить в Интернет через смартфон;
- с лонгридом вы сможете рассказать историю: рассказать о себе, своих целях и специфике работы, вызвать эмоции — все это поможет получить новые партнерские предложения, а также новых ценителей бренда;
- лонгридами очень любят делиться в социальных сетях;
- формат такой статьи способствует продолжению общения, установлению контакта;
- с таким материалом вы сможете рассказать о своем бизнесе максимально завуалированно, чего не скажешь, например, о лендинге;
- и, наконец, хороший лонгрид действительно привлекает трафик и увеличивает конверсию, помогает набрать просмотры, лайки и репосты, а также повысить доверие аудитории.

Обязательное условия такого текста — это возможность полностью погрузить читателя в текст, который он прочитает от начала до конца. Есть приемы, которые позволяют создать такой качественный материал, способный собрать множество лайков.
- Понятный язык. Не оставляйте в тексте терминов и фраз, которые могут вызвать непонимание у читателя, тем самым выбив его из “глубокого” чтения.
- Детали, цепляющие внимание. Для того, чтобы читатель не заскучал и получал информацию в разнообразных формах, разбавьте текст своеобразными “якорями” вроде цитат, вопросно-ответной формы и других интересных вариантов.
- Мультимедийные файлы. Для того, чтобы чтение по-настоящему затянуло, нужно применять в работе всевозможные варианты визуализации, которые будут хорошо сочетаться с текстом.
- Повысьте доверие.
 Для того, чтобы человек видел, что вся представленная информация не является ложной, необходимо снабдить материал гиперссылками на источники: иностранные и авторитетные исследования, которые использовались для работы.
Для того, чтобы человек видел, что вся представленная информация не является ложной, необходимо снабдить материал гиперссылками на источники: иностранные и авторитетные исследования, которые использовались для работы.
Написание такого текста, как лонгрид, требует большой подготовки и много часов работы: нужно собрать максимум материала и суметь структурировать его, убрав лишнее, сохранив при этом объем. Научиться писать длинные тексты, которые будут захватывать внимание, можно на курсах журналистики, где вы обучитесь всем секретам работы с такими материалами.
Поделиться статьёй:
NIT for You | Лонгрид: понятие и построение
Лонгрид (от англ. “long read” – длинное чтение) – это принципиально новый способ подачи объемного текста в Интернете. Также этот формат называют deep read («глубокое чтение»), и второе определение куда ближе по смыслу.
Качественный лонгрид – отнюдь не километровая простыня текста, а продуманная статья, раскрывающая тему максимально полно. Выделим основные принципы.
Выделим основные принципы.
Принцип мультисенсорного опыта
Лонгрид должен восприниматься как целостная история. При этом он интересен только в том случае, если он будет прямо-таки «фонтанировать» интересными фактами, яркими цитатами, малоизвестными деталями.
Среди пользователей Интернета закрепился минимальный объем публикации – 1500 знаков, хотя их количество не ограничено и определяется глубиной разработки темы.
Сторителлинг позволяет не только узнать о чем-то, но и испытать это на себе: видео, аудио, типографика и анимация создают атмосферу и позволяют получить цельное впечатление от истории.
Принцип скимминга (избирательное чтение)
При избирательном чтении мы обращаем внимание прежде всего на картинки и подписи, и visual storytelling создан как раз для такого образа чтения. Правильная расстановки визуальных акцентов позволяет усваивать информацию быстро и комфортно.
Лонгрид оформляется как отдельный сайт, со своей уникальной версткой, дизайном, что опять-таки отличает его от обычной статьи. Как известно, длинный текст в сети не приветствуется, ведь зачастую в Интернет люди приходят получить информацию быстро и в максимально сжатые сроки. Но лонгрид – дело совершенно другое. Это не сплошной массив текста, а целый комплекс, состоящий из текста, разбавленного видеороликами, инфографикой, презентацией, фоновой музыкой, иллюстрациями (в том числе интерактивными), сносками, встроенными цитатами и т. д. Самое важное, что все они взаимосвязаны между собой − они не просто дополняют текст, а являются его равноправными партнерами. В таком материале крайне важна последовательность изложения, гармоничное сочетание всех его частей – размер и количество иллюстративного ряда, шрифтов, цветов и другое.
Как известно, длинный текст в сети не приветствуется, ведь зачастую в Интернет люди приходят получить информацию быстро и в максимально сжатые сроки. Но лонгрид – дело совершенно другое. Это не сплошной массив текста, а целый комплекс, состоящий из текста, разбавленного видеороликами, инфографикой, презентацией, фоновой музыкой, иллюстрациями (в том числе интерактивными), сносками, встроенными цитатами и т. д. Самое важное, что все они взаимосвязаны между собой − они не просто дополняют текст, а являются его равноправными партнерами. В таком материале крайне важна последовательность изложения, гармоничное сочетание всех его частей – размер и количество иллюстративного ряда, шрифтов, цветов и другое.
Лонгрид : чем поможет в учебном процессе?
Типы лонгридов
На сегодняшний день принято выделять несколько типов лонгридов:
- Репортаж.
 Раскрывает все подробности события (чаще всего спустя время после него).Фишки, которые можно использовать:
Раскрывает все подробности события (чаще всего спустя время после него).Фишки, которые можно использовать:- аудиовставки;
- анимированный фон;
- интервью с участниками;
- видеонарезки с мероприятия.
- Реконструктор. Этот материал является реконструкцией цепи каких-то определенных событий, попыткой проанализировать их, выяснить причины и последствия. В таком типе лонгрида крайне важную роль играют дополнительные эффекты: фоновая музыка, видеовставки – именно они задают нужное настроение при прочтении.
- Портреты (второе название − people stories). К этому типу относятся и портретные интервью, и очерки, и биографические зарисовки. Главная задача – раскрыть личность героя, его жизненные принципы, взгляды. Фишки, которые можно использовать:
- цитаты героя статьи и его близких;
- фотографии;
- уникальные факты, которых нигде нет.
- Коммерческие лонгриды – это не рекламные материалы, не лендинги, не продающие тексты.
 Они предназначены для того, чтобы предоставить постоянным и потенциальным клиентам исчерпывающую информацию о компании, о том, как производится продукт, о самом продукте, о тенденциях сферы деятельности компании.
Они предназначены для того, чтобы предоставить постоянным и потенциальным клиентам исчерпывающую информацию о компании, о том, как производится продукт, о самом продукте, о тенденциях сферы деятельности компании. - Мультимедийный лонгрид. Это – статьи, щедро разбавленные картинками, анимацией, яркими надписями, видеороликами и т.д. Собственно, любой из лонгридов можно отчасти отнести к этому типу, однако здесь есть один важный нюанс. Как правило, в лонгридах главную роль играет текст – остальной «декор» его только дополняет. В мультимедийной же версии все детали равноправны, а иногда текст идет связующим звеном между графическими элементами.
Основные шаги построения
Инструкция: Как писать и оформлять лонгрид: ссылка
Кроме того, не стоит забывать и про две основные модели потребления контента современным читателем: leanback (читают, откинувшись назад) и leanforward (наклонившись вперед). Если новости, короткие видео и небольшие заметки относятся к последнему типу, то лонгриды ложатся в одну категорию с кинофильмами, сериалами, журналами и книгами – их читают, удобно расположившись в кресле или на диване.
Источники:
- https://tilda.cc/ru/longreads/
- http://vashredaktor.com/chto-takoe-longrid/
- https://internet-marketings.ru/kak-sozdat-longrid/
- http://madcats.ru/content-marketing/longreads-how-to/
- http://wordfactory.ru/prodayushhij-longrid-glavnyj-format-kommercheskogo-teksta/
- https://geniusmarketing.me/lab/kak-napisat-longrid/
- http://mediatoolbox.ru/longread/
- https://tjournal.ru/stories/93040-instrukciya-kak-pisat-i-oformlyat-longrid
Что такое «Лонгрид» и с чем его едят?
В современном мире веб-дизайна и интернет-разработок лонгридом можно считать как мультимедийное ответвление от Лендинг Пейдж, посадочной страницы.Каждый день в интернете создают множество различных терминов. Это касается и текстов, которые размещаются в сети. Если вы копирайтер, который держит руку на пульсе событий, вы должны быть в курсе современных трендов в копирайтинге. Даже если какое-то профессиональное понятие не на слуху, вы должны знать, что оно означает. К примеру, знаете ли вы, что такое «лонгрид»? Вы могли часто слышать этот термин, но, как показывает практика, не все знают его точное значение.
К примеру, знаете ли вы, что такое «лонгрид»? Вы могли часто слышать этот термин, но, как показывает практика, не все знают его точное значение.
Лонгрид (от англ. “long read” – длинное чтение) – это абсолютно новый инструмент создания объемных текстов для сетевых ресурсов. Мы все знаем, что длинные тексты не слишком популярны в сети. Людям нужна четкая информация – максимально понятная и без воды. Но лонгрид – это универсальный способ донести много текста пользователю и он его прочитает! Т.е. в современном мире веб-дизайна и интернет-разработок лонгридом можно считать как мультимедийное ответвление от «Лендинг Пейдж» (посадочной страницы).
Секрет лонгрида в том, что это не сплошная простыня текста, а так называемый комплекс подачи информации. Текстовые блоки в лонгридах разбавляют изображениями, видеороликами, инфографикой, презентациями, музыкой – да всем чем угодно. Интерактивные элементы в таком тексте – это не просто замыливание глаз, а полноценная часть изложения. К примеру, вы пишете про кошачий корм и во время повествования, продолжаете строчку о его вкусовых качествах роликом, на котором кот с удовольствием уплетает рекламируемый продукт, после ролика вы продолжаете свой сео-текст без потери пользовательского внимания!
Увлекая читателя разнообразной подачей материала, вы уже становитесь не просто копирайтером написавшим текст для поисковика. Вы создаете настоящую аналитику, которая подана в формате увлекательного рассказа.
Существует три основных типа лонгридов:
- Репортаж. Тут главное реалистично рассказать о предоставляемой услуге. Повествование должно вестись от третьего лица, но старайтесь избегать употребления в повествовании слова «я».
- Реконструкция. Это настоящая аналитика. Вам необходимо описать и проанализировать цепь определенных событий, которые приведут к необходимому результату. В реконструкции важную роль играют музыкальное сопровождение, видеоряд и графический контент.
- Портреты (второе название ? people stories) как вы уже догадались портретные лонгриды – это отзывы, интервью, биографические зарисовки и очерки.
 Во время повествования копирайтеру необходимо раскрыть личность героя, рассказать о нем пользовательской аудитории.
Во время повествования копирайтеру необходимо раскрыть личность героя, рассказать о нем пользовательской аудитории.
Как правильно создать лонгрид?
- Лонгрид – очень сложный структурированный материал. Если вы только-только вступили на стезю журналистики/копирайтинга – лонгрид не ваш вариант. Для него вам необходимо набраться опыта.
- Изучите тему, которую собираетесь освещать! Не начинайте работу с наскока. Изучите материалы по теме, ознакомьтесь с различными мнениями, используйте в своей работе разные источники информации. Завлеките своего читателя интересной подачей, редкими фактами и запоминающимися видеографическими материалами.
- Расставьте акценты и составьте четкую схему того, о чем вы собираетесь писать. Лонгрид – это не художественный роман, а структурированный аналитический материал.
- Перед началом работы – сделайте для себя примерный набросок того, как будет выглядеть ваш лонгрид.

плюсы использования лонгридов для дистанционного обучения.
Лонгрид становится одним из основных трендов в e-learning. Главное преимущество long read в том, что он позволяет сделать так, чтобы большой объем текста не пугал обучающегося, а вовлекал. Вовлечь в чтение и изучение информации помогают мультимедийные элементы: изображения, видео, графика.
Такие элементы наглядно разъясняют и дополняют текст, усиливают эмоцию, помогают выразить суть, которую сложно отобразить словами.
Кроме этого, отдельные текстовые части в лонгриде можно выделить с помощью типографики:
- примечания,
- пояснения, сноски,
- факты,
- перечисления,
- цитаты,
- блоки внимание.
В результате упрощается восприятие и запоминание обучающей информации. Если в текст добавить яркие примеры, то процесс обучения станет еще более интересным и увлекательным.
Еще одно достоинство лонгрида как средства обучения в том, что в него можно поместить большой объем обучающей информации. Например, создавая слайдовые курсы, мы делаем полезную выжимку из нескольких нормативных документов, помещая на слайды только основную информацию, ключевые мысли и краткие выводы.
Работая над созданием лонгридов, мы также перерабатываем исходную информацию, полученную от заказчика, с той разницей, что у нас появляется возможность добавить в обучающий материал больше примеров и пояснений.
Какой материал можно изложить в формате лонгрид
В формате лонгрид можно изложить практически любую обучающую информацию. Это может быть разъяснение локально-нормативных актов, которые регламентируют трудовую деятельность компании и персонала, например: регламенты, инструкции, руководства, положения и др.
Сегодня в связи с массовым переводом предприятиями сотрудников на дистанционный режим работы в домашних условиях, становятся актуальны лонгриды на такие темы, как:
- Политика безопасности при работе на дому
- Правила организации эффективных онлайн встреч
- Тайм-менеджмент
- Адаптация в новых условиях работы
- Руководство удаленными сотрудниками и др.
Большое внимание при создании лонгрида мы уделяем структурности, ясности и полноте информации. Как правило, обучающий материал делится на главы, подразделы и логические блоки. Обучающийся при изучении материала имеет возможность ознакомиться с дополнительными примерами, пояснениями или видео к курсу.
Кроме этого, мы стремимся сделать текстовый материал визуально красивым, интересным и захватывающим. По желанию заказчика мы добавляем уникальные сюжеты, сценарий и примеры, чтобы обучающийся вовлекался в чтение, и ему было проще запомнить информацию.
Например, в лонгриде могут быть приведены примеры решений сложных задач командой, мнение экспертов вашей отрасли, результаты исследований по теме обучения, прогноз или статистика.
Завершаться курс может тестированием для проверки знаний. Анализируя ответы, вы сможете оценивать прогресс обучающихся, а также проводить работу над ошибками: давать пояснения, добавлять полезные факты и др.
Заказать разработку лонгрида
Компания «Дискус» имеет успешный опыт разработки лонгридов и слайдовых курсов для обучения сотрудников. Мы знаем, как сделать процесс обучения интересным и захватывающим.
Мы знаем, как сделать процесс обучения интересным и захватывающим.
По вашему заданию наши методисты переработают нормативные документы или материал ваших авторов (руководителей, тренеров, педагогов), составят сценарий и структурируют информацию.
Затем наши дизайнеры разработают концепцию и стилистику дизайна, подберут визуальный контент, а технические специалисты разместят лонгрид в Вашей СДО. Мы используем адаптивную верстку лонгрида, чтобы он корректно отображался на всех устройствах, которые могут использоваться обучающимися (компьютер, ноутбук, планшет, смартфон).
Если перед вами стоит задача по обучению сотрудников, партнеров или руководителей, то обращайтесь в нашу компанию. Мы сможем порекомендовать оптимальный формат для обучающего курса и ответим на все ваши вопросы.
Посмотрите Наше Портфолио и Готовые курсы
Заявка на презентацию
Мультимедийный лонгрид как новый журналистский формат Текст научной статьи по специальности «СМИ (медиа) и массовые коммуникации»
журналистика. К новостным постам часто прикрепляются интернет-мемы и инфографика.Социальные медиа находятся в тесной связи с профессиональными журналистскими платформами региона. Социальная сеть «ВКонтакте» — самая популярная социальная площадка в России — выступает и как платформа конкурентной борьбы интернет-СМИ, и как самостоятельный проект гражданской журналистики.
К новостным постам часто прикрепляются интернет-мемы и инфографика.Социальные медиа находятся в тесной связи с профессиональными журналистскими платформами региона. Социальная сеть «ВКонтакте» — самая популярная социальная площадка в России — выступает и как платформа конкурентной борьбы интернет-СМИ, и как самостоятельный проект гражданской журналистики.
В сообществах официальных СМИ республики Коми в борьбе за интерес читателей ключевым звеном выступают прямой интерактив с подписчиками и публикация присылаемой от них в редакцию мультимедийной информации. Немаловажным остается факт, что издания за счет взаимного информационного обмена с читателем получают первые подсказки к горячим новостям и общественным резонансам. Сегодня такие сообщества — лишь вспомогательное средство для перехода пользователя из соцсети на сайт издания за счет ссылок, использования хештегов и визуальных жанров. Последние представлены статичными и художественными фотоиллюстрациями, фоторепортажами, видеоиллюстрациями и инфографикой.
Существенным недостатком сетевых СМИ региона, тем не менее, остается их почти полная подконтрольность властям, как следствие — отсутствие злободневной общественно-политической тематики и реальной
конкуренции между различными ресурсами. В связи с этим можно отметить, что на фоне стремительного развития мультимедиа в центральных СМИ (интерактивные карты, «сторителлинг», «таймлайн»), республиканские сайты используют традиционные формы фото- и видеоиллюстрирования своих материалов, а различные проекты развития местных сетевых радио- и телеканалов так и не были реализованы. Однако логика прогресса в области интернет-коммуникаций подсказывает, что их появление — дело неизбежное. Возможно, недавние коррупционные скандалы в правительстве республики и обнародование схем подавления свободы слова в региональных СМИ будут способствовать оживлению медиасферы Коми и коснутся в том числе поисков новых аудиовизуальных форм подачи информации.
Литература
1. Пуля В. 7 трендов: что ждет новые медиа в 2014 году? // Журналист. — 2014. — №1. — С. 48-49
2. Калмыков А.А., Коханова Л.А. Интернет-журналистика. М., 2005.
3. Бугаева И. Демотиваторы как новый жанр в Интернет коммуникации, жанровые признаки, функции, структура, стилистика. 2011. [Электронный ресурс] / URL: http://www.rastko.rs/filologya/ stil/2011/10Bugaeva.pdf
УДК 070:002
М. Н. Булаева
Южно-Уральский государственный университет (НИУ)
МУЛЬТИМЕДИЙНЫЙ ЛОНГРИД
КАК НОВЫЙ ЖУРНАЛИСТСКИЙ ФОРМАТ
Влияние интернет-технологий на журналистику существенным образом отразилось на ее содержании. Появились новые форматы представления информации в интернете, одним из которых является мультимедийный лонгрид. Это интерактивный материал, созданный на основе объемного текста и содержащий аудиовизуальные компоненты. В данной статье формулируется понятие мультимедийного лонгрида, дается его общая характеристика, описываются содержательные и формальные признаки, исследуются композиционные особенности.
Появились новые форматы представления информации в интернете, одним из которых является мультимедийный лонгрид. Это интерактивный материал, созданный на основе объемного текста и содержащий аудиовизуальные компоненты. В данной статье формулируется понятие мультимедийного лонгрида, дается его общая характеристика, описываются содержательные и формальные признаки, исследуются композиционные особенности.
Ключевые слова: журналистика, СМИ, мультимедийный лонгрид, формат.
The influence of internet technology upon journalism had a huge impact on its content. New information formats such as multimedia longread appeared. It is an interactive material based on a long text with audiovisual components in it. The article represents a multimedia longread concept, gives its general characteristics, describes substantial and formal features, along with its compositional peculiarities.
Keywords: journalism, mass media, multimedia longread, format
ВОПРЕКИ РАСПРОСТРАНЕННОМУ МНЕНИЮ о том, что длинные тексты в интернете не востребованы аудиторией, на практике можно убедиться в обратном. Современные технологии представления контента позволяют адаптировать объемные материалы для интернет-среды. Одним из таких успешных журналистских форматов является мультимедийный лонгрид («long read» — длинное чтение) — это «новый формат подачи текстовой информации в интернете» [1], в основе которого — текст большого объема и аудиовизуальные компоненты. Обычно это подробный обзор по теме, содержащий помимо текста различные мультимедийные элементы: фото, видео, инфографику и т.п. Таким образом, можно охарактеризовать лонгрид как мультимедийный проект, посвященный конкретному событию или проблеме, актуальной дате. Ключевой характеристикой такого формата является объем и продолжительность материала, а основой — журналистский текст. Лонгриды обычно создаются на отдельной
122
Журналистский ежегодник
странице и имеют особую верстку: мультимедийные элементы и основной фон движутся с разной скоростью (эффект параллакса).
Для обозначения конкретного формата слово «лон-грид» стало широко использоваться после выхода мультимедийного проекта The New York Times «Snow Fall» в 2012 году, завоевавшим огромную популярность у аудитории и в журналистской среде. Сегодня лонгриды стали уже привычным явлением в зарубежных СМИ (The New York Times, The Guardian, The Chicago Tribune, The Seattle Times и др.). Растет популярность формата и в российской журналистике. Мультимедийные лонгриды создают МИА «Россия сегодня» (цикл мультимедийных статей «15 ударов Красной армии»), Lenta.ru («Дни затмения», «Конец советов 20 лет»), «Коммерсантъ» («Земля отчуждения», «Приразломная жизнь»), «Комсомольская правда» («Америка заколоченных небоскребов»), «Первый канал» («Всем миром. Год спустя») и др. Развитие технологий позволяет упростить процесс создания лонгри-дов, формат становится доступным даже для небольших редакций. Так, для верстки лонгридов существуют готовые онлайн-платформы tilda. cc, creatavist.com, medium. com и другие, а также мобильные приложения Adobe Slate, Storehouse, Pixotale.
cc, creatavist.com, medium. com и другие, а также мобильные приложения Adobe Slate, Storehouse, Pixotale.
Исследователь А.В. Колесниченко рассматривает лонгрид как жанр журналистики. Он отмечает, что его главной жанровой характеристикой является системность темы (новое явление, системное расследование), глубокое и длительное исследование темы журналистом с использованием большого количества источников информации и большой объем текста в сочетании с высокой плотностью смысла и претензией на исчерпанность данной темы [2]. На наш взгляд, в качестве текстовой основы лонгрида могут выступать такие жанры, как репортаж, статья, очерк, обозрение, журналистское расследование и их элементы. В совокупности с мультимедийными платформами они формируются в определенную структуру, в знаковых системах интернета приобретают новые формы и дают возможность характеризовать лонгрид как формат универсальной журналистики. В отличие от жанра, суть которого определяется содержательными характеристиками материала, формат характеризует, прежде всего, структурные особенности медиапродукта.
Редактор спецпроектов ИД «Коммерсантъ» А. Галустян, говоря о тематических особенностях формата мультимедийного лонгрида, отмечает, что «он дает зрителю ощущение присутствия и переживания. Это означает, что тема должна быть общедоступной, должна касаться многих и быть интересной, но при этом в нее должно быть сложно окунуться. Такой проект должен отправлять зрителя туда, где он не сможет по каким-то причинам оказаться» [3]. Лонгрид позволяет создать яркие визуальные образы, раскрыть драматургию события, динамично описать процессы. Такие материалы отличаются особой структурой: композиция текста строится по схеме параллельного рассказа, смены повествова-
ния и объяснений, наличия историй и репортажных фрагментов. При просмотре лонгрида аудитория не только читает текст, но и одновременно знакомится с остальными элементами: видео-, аудиозаписями, 360-градусными панорамами, интерактивной графикой, инфографикой, фотогалереями, слайд-шоу. Аудиовизуальные элементы лонгрида распределяются по всему материалу, некоторые из них можно просмотреть по ходу чтения, а некоторые открываются только при обращении к ним. При этом аудитория сама определяет порядок просмотра элементов. Поэтому наиболее важной задачей, стоящей перед авторами мультимедийного лонгрида, является создание единого целостного материала, включающего в себя разнообразные мультимедийные элементы.
Аудиовизуальные элементы лонгрида распределяются по всему материалу, некоторые из них можно просмотреть по ходу чтения, а некоторые открываются только при обращении к ним. При этом аудитория сама определяет порядок просмотра элементов. Поэтому наиболее важной задачей, стоящей перед авторами мультимедийного лонгрида, является создание единого целостного материала, включающего в себя разнообразные мультимедийные элементы.
Для лонгрида характерен особый способ построения материала, который можно охарактеризовать как мультимедийное нарративное изложение («письменное повествование»). Нарративное изложение — это явление, сочетающее в себе свойства журналистики и художественной литературы: с одной стороны, оно затрагивает социально значимые темы, отличается фактографической точностью излагаемой информации; с другой стороны, представляет увлекательные истории, написанные живым языком, с драматическим сюжетом, в которых явно звучит голос автора [4].
Мультимедийное нарративное изложение обладает интерактивностью, которая обеспечивает активную роль аудитории (пользователей) при определении порядка и количества просмотренных элементов. Понятие «мультимедийное нарративное интерактивное изложение» как способ построения материала в формате лонгрида мы понимаем как повествование, посвященное актуальной социально значимой теме, имеющее драматическую структуру, сочетающее в себе свойства журналистики и художественной литературы, созданное на основе сочетания различных медиаплатформ (текст, фото, видео, аудио, графика, инфографика, анимация) и предполагающее такое взаимодействие аудитории с материалом, при котором она самостоятельно осуществляет выбор количества и порядка просмотра всех его элементов.
Таким образом, мультимедийный лонгрид — это особый формат передачи информации в интернете, основой которого является журналистский текст, создающийся на отдельной странице; отличающийся большим объемом и продолжительностью материала во времени; сочетающий в себе все многообразие аудиовизуальных, графических средств; основанный на мультимедийном интерактивном нарративе. Это специальный коллективный мультимедийный проект редакции, посвященный конкретному событию, важной проблеме, актуальной дате, многоаспектной теме, предполагающей максимально полное изучение материала.
Это специальный коллективный мультимедийный проект редакции, посвященный конкретному событию, важной проблеме, актуальной дате, многоаспектной теме, предполагающей максимально полное изучение материала.
Для успешного создания лонгрида в интернете универсальный журналист должен обладать всеми необходимыми личностно-профессиональными качествами, технологическими навыками и умениями, а также иметь
«навык видения мультимедийного замысла» [5. С. 65], позволяющий ему уже на подготовительном этапе представить конечный продукт.
Литература
1. Что такое лонгрид? // Современная библиотека // URL: http:// sbibli0teka.bl0gsp0t.ru/2014/11/bl0g-p0st_57.html (дата обращения: 14.09.2015)
2. Колесниченко А.В. Длинные тексты (лонгриды) в современной российской прессе / А. В. Колесниченко // Медиаскоп. — 2015. — №1 // URL: http://www.mediasc0pe.ru/n0de/1691 (дата обращения: 11.09.2015)
3. Колотилов В. Интерактивные статьи, которые учат по-новому рассказывать истории / В. Колотилов // URL: http://med iakritika. by/article/2212/interaktivnye-stati-k0t0rye-uchat-p0-n0V0mu-rasskazyvat-ist0rii (дата обращения: 22.09.2015)
4. Бозрикова С.А. История нарративной журналистики в России / С. А. Бозрикова // URL: http://www.academia.edu/3684620/ (дата обращения: 14.09.2015)
5. Медиаконвергенция и мультимедийная журналистика: Материалы к обучающим семинарам / Сост. С. Балмаева. — Екатеринбург: Изд-во Гуманитарного университета, Кабинетный ученый, 2011. — 148 с.
УДК 398.2+316.776 (+13)
Т. И. Суслова
Томский государственный университет систем управления и радиоэлектроники
ИНТЕРНЕТ-ФОЛЬКЛОР КАК СРЕДСТВО КОММУНИКАЦИИ
Феномен интернет-фольклора в статье рассматривается как одна из ярких форм современной коммуникации, своеобразного медийного общения. Автор утверждает, что бытование фольклора в веб-пространстве позволяет сохранять основные традиционные ценности культуры не в качестве музейных реликтов, а живых и подвижных форм современной художественной практики и культуры. Конструирование, изобретение традиций усиливается с возникновением новейших информационных технологий, человек же обращается к реконструкции прошлого для обретения новых опор бытия в реальном мире через сетевое творчество.
Ключевые слова: интернет, фольклор, традиции и инновации, симулякр, сетевое сообщество.
The phenomenon of Internet- folklore article regarded as a form of modern aesthetic consciousness. The author argues that folklore in the web- space allows you to store basic traditional values of the culture is not as museum relics, a live and mobile forms of contemporary artistic practice and culture. Design, invention of tradition is enhanced with the emergence of new information technologies, and man is drawn to the reconstruction of the past to support the acquisition of new life in the real world through a network creative work.
Keywords: Internet- folklore, tradition and innovation, simulacrum, society of network.
ГЛОБАЛИЗАЦИЯ КАК ТЕХНИЧЕСКИЙ ПРОЦЕСС, с одной стороны, способствует гомогенизации, становлению единого мира, с другой, — обостряет в силу усиления интенсивности и глубины межкультурного общения проблему межкультурных различий и культурной экспансии. Современные средства коммуникации — от Интернета до транспортных средств и новейших технологий, создающих мир комфорта, не вызывают неприятия как
технико-экономический процесс взаимодействия стран и народов, пока речь не идет о культурной составляющей этого взаимодействия. При этом усвоение английского компьютерного сленга, авто- и компьютерной техники Японии представителями иных языковых групп само по себе не способствует сохранению, распространению или экспансии, например, английской или японской культур. Согласно трактовке ряда исследователей, современная ситуация в культуре рассматривается как совокупность альтернативных вариантов развития, имеющих три подсистемы: фазы атрофии, стабилизации и возникновения. В первой преобладают символика и традиции культуры прошлого; вторая является несущим элементом культуры; третья вырабатывает новый тип символических культурных связей. Интернет-культура в фольклорных формах предстает как одна из ярчайших субкультур современности, которая синтезирует все представленные варианты развития современной культуры и вызывает наибольший интерес. Революционные темпы эволюции технических средств, формирования сознания человека, развития демократических институтов направлены на усиление темпов развития социального порядка, в котором каждый человек и культурный феномен обретают значимость как системы. Период трансформаций — время ускорения темпов жизни, вписывание в которые порождает образцы жизни «на скорую руку» или «инстант-культуру». Она основывается на «быстром знании» или смыслах, в сущности представляющих стереотипы, архетипы, которые предлагают образцы, цели и способы достижения целей коротким путем. Согласно таблице Плятта-Шкловско-го-Пряхина, сегодня достигнуты определенные пределы
долго читаемых сообщений в блогах: 10 советов по привлечению читателей | StoryChief
Посмотрите видео для TL; DR
Недавний опрос Orbit Media показал, что блоггеры, которые тратят больше времени на написание своих блогов, а также пишут более длинные статьи, обычно имеют большее количество читателей и взаимодействий. Лучшие результаты SEO достигаются в блогах, состоящих примерно из 2000 слов. Однако как сделать так, чтобы долгое чтение было интересным? Как сделать так, чтобы ваши читатели не просто пролистывали или не нажимали, когда они прочитали половину вашей статьи?
Блогеры, которые тратят 6 и более часов на публикацию, на 56% чаще сообщают о «хороших результатах», чем те, кто этого не делает.(Джоди Харрис, Институт контент-маркетинга)
Вот несколько полезных советов, которые значительно улучшат вашу долго читаемую игру.
Оглавление:
1. Добавьте видео в свои блоги, которые читают долго 4. Что это, кроссовер? (пригласите приглашенного писателя)
5. Один для дороги (адаптируйте свой длинный текст для мобильных устройств)
6.Пишите, отслеживайте, измеряйте, изменяйте (учитесь у аналитиков)
7. Ищите редактора
8. Разнообразие — это приправа жизни (и долгое чтение)
9. Освежите память ваших читателей (используйте якорные ссылки)
10. Пишите умнее, а не сложнее (тратить меньше времени на публикацию)
1. Добавляйте видео в свои давно читаемые блоги
Одна из самых популярных тенденций в блогах 2019 года — добавлять видео в свои блоги. Люди обычно просто любят смотреть видео. Это делает их отличным дополнением к созданию вашего контента, поскольку люди дольше «задерживаются» на вашем веб-сайте, и поэтому Google признает ваш блог ценным.
С помощью нашего инструмента вы можете легко разместить видео в своем блоге.
2. Обеспечьте ваше длинное чтение достаточным количеством наглядных материалов
По словам Аннелин Офофф из Vranckx, читатели в два раза чаще запоминают информацию, когда она представлена визуально. Ваша история значительно выиграет от использования идеального визуального элемента, поддерживающего ваш текст. Если сообщение в блоге содержит только текст, люди могут немедленно щелкнуть мышью. Сделайте так, чтобы ваше длинное чтение выглядело как красочное путешествие, а не тяжелый русский роман.
Фестиваль Холи, автор: AJP / ShutterstockЧем больше чувств может задействовать визуальный элемент, тем больше внимания он получает и тем больше информации сохраняется. (Новости)
Теперь вы можете легко добавлять изображения в нашем редакторе благодаря новой интеграции Unsplash. Unsplash — поставщик стоковых изображений, предназначенный для бесплатного обмена фотографиями. В их библиотеке более 800 000 фотографий, так что даже самые придирчивые блоггеры могут найти идеальное изображение для своей статьи.
3.Сначала напишите длинное чтение для людей, а затем структурируйте его для поисковых систем
Как мы уже упоминали, когда ваш блог содержит около 2000 слов, он с большей вероятностью окажется выше в результатах поиска Google. Однако это не означает, что вы должны стараться сделать каждый блог длиной 2000 слов. Если тема вашего блога не поддается более длинному формату, нет смысла растягивать ее, пока вы не достигнете отметки в 2000 слов. Только если ваше сообщение лучше всего передается с помощью длительного чтения, вам следует подумать о том, чтобы написать его.
4. Что это, кроссовер?
Обновите свой длинный пост в блоге, добавив новое лицо. Большинство людей уже знают, что наличие в вашем блоге приглашенного писателя — отличный способ привлечь посещаемость вашего блога. Так почему бы не сотрудничать с кем-нибудь из вашей сети, у кого есть ценная информация по теме, которую вы хотели бы затронуть? Сочетание стилей привносит в вашу статью глоток свежего воздуха. Более того, результаты опроса Orbit Media показывают, что наличие в вашем блоге приглашенного писателя значительно увеличивает количество читателей.
Блогеры, которые публикуют гостевые сообщения, на 48% чаще сообщают о хороших результатах, чем блогеры, которые этого не делают. (Энди Крестодина, Orbit Media)
В редакторе StoryChief вы можете легко пригласить приглашенных авторов в свой блог, добавив их в качестве пользователей (с подкатегорией «приглашенный автор»), а затем пригласив их в свою статью.
Нужна обратная связь? Спросите своих коллег или редактора!5. Один для дороги (адаптируйте свой лонгрид для мобильного телефона)
У большинства из нас напряженная жизнь, не так ли? А иногда бывает очень удобно иметь возможность продолжить чтение длинного сообщения в блоге, пока вас нет дома или в дороге.Убедитесь, что вы даете своим читателям свободу читать, где и когда они хотят. Не привязывайте их к одному месту, пусть они резвятся по миру с вашим блогом в руках.
В нашем редакторе истории автоматически адаптируются ко всем экранам, поэтому вам больше не нужно об этом беспокоиться.
Сначала мобильные, потом баскетбол.6. Записывайте, отслеживайте, измеряйте и изменяйте свое длинное чтение
Практика делает совершенство. Нет ничего плохого в том, чтобы создать плохой пост в блоге. Однако не учиться на своих ошибках в долгосрочной перспективе может быть вредно.Вы не всегда можете знать, будет ли ваш пост в блоге успешным или нет, но вы можете многому научиться из своих прошлых результатов.
Аналитика помогает вам лучше понять, что действительно ценит ваша аудитория, вместо того, чтобы полагаться на собственное предвзятое мнение о том, какой контент является хорошим. Более качественный контент в сочетании с лучшим пониманием того, что ценит ваша аудитория. — ключ к повышению производительности. (Джон Холл, Calendar.com)
С нашим редактором вы никогда не будете полностью в темноте, когда дело доходит до предвидения будущего вашей статьи.Он позволяет вам проверить, сколько людей нажали на вашу статью и сколько людей на самом деле прочитали ее.
7. Ищите редактора
Большинство писателей знают, что перечитывание — самая важная часть письма. Многие из них также знают, что может быть удобно позволить сторонним лицам просмотреть ваши статьи. Мы все были там и знаем, как легко упустить ошибку. Решите эту проблему, попросив другого человека проверить ваш текст перед публикацией.
С помощью нашего инструмента вы можете просто пригласить гостя оставить отзыв о вашем блоге! Затем этот гость может оставлять комментарии, чтобы сообщить вам, где вы допустили ошибку, или где вы могли бы сформулировать предложение немного ярче.
Две головы умнее одной.Однако иногда вы просто не хотите просить другого человека просмотреть ваш контент. И да, мы тоже об этом думали! Наш редактор проверяет читабельность вашей статьи, что является очень важным аспектом онлайн-написания (если вы не пытаетесь переписать «Улисс» Джеймса Джойса, вы, вероятно, вообще не хотите, чтобы ваш текст был читабельным). Он также проверяет, полностью ли оптимизировано SEO вашей статьи.
8. Разнообразие — это изюминка жизни (и длинные чтения)
Важно заставить людей читать ваши длинные книги, но превращение этих читателей в постоянных клиентов может быть для вас еще более ценным.Когда вы стремитесь вести блог для своей компании, важно привязать клиентов к вашему бренду. Лучше всего это делается по старинке, часто публикуя. Результаты опроса Orbit Media показали, что люди, которые публикуют более одного блога в неделю, имеют больше шансов получить хорошие результаты.
(Источник: Orbitmedia.com)Мы не говорим, что вы должны публиковать одно длинное сообщение в блоге за другим, потому что оно может быстро устареть. Тем не менее, если время от времени проводить долгое чтение, это может значительно улучшить ваши общие результаты.
9. Освежите память читателей
Если вы хотите, чтобы ваш текст был еще проще читателям, вы можете добавить якорные ссылки. С помощью якорных ссылок вы можете ссылаться на более ранние части вашего текста, что, в свою очередь, значительно улучшит восприятие вашей аудитории. Им просто нужно щелкнуть ссылку, и они в мгновение ока могут вспомнить, о чем вы писали ранее в тексте. На первый взгляд это может показаться небольшим дополнением, но будет больше шансов, что ваши читатели действительно вернутся, чтобы перечитать предыдущую часть с якорной ссылкой.Это также поможет им лучше понять ваш блог в долгосрочной перспективе, поскольку ключ к запоминанию — это повторение.
С помощью редактора StoryChief вы можете легко реализовать якорные ссылки. Выберите слово, фразу или предложение. Нажмите на значок ссылки, как если бы вы это делали, когда хотите добавить внешние ссылки. Измените раскрывающийся список с «внешнего» на «источник». Затем просто выберите часть вашего текста, к которой вы хотите вернуться.
10. Работайте умнее, а не усерднее
Мы не собираемся лгать: если вы хотите, чтобы ваш блог был успешным, вам нужно много работать.Наилучшие результаты видят люди, которые пишут длинные статьи, тратят более 6 часов на их создание и публикуют чаще одного раза в неделю. Если вы действительно серьезно относитесь к улучшению своей контент-стратегии, вы должны быть готовы сделать это существенной частью своей недели. Вот почему наши ленивцы в StoryChief делают все возможное, чтобы упростить вам ведение блога и максимально увеличить время написания. Не тратьте время на бюрократию. Получите максимум от своего времени, потратив его на творчество и самосовершенствование.
И не забывайте … Публикуйте ваши лонгриды многоканально!
Если вы приложили все усилия к созданию сообщения в блоге, вы, вероятно, захотите, чтобы его прочитало как можно больше людей. Чем больше каналов публикуется в вашем блоге, тем чаще его будут замечать в Интернете. Это может быть довольно утомительно, но наш редактор предлагает вам возможность публиковать материалы на нескольких каналах одним щелчком мыши. Написание долго читаемой истории само по себе занимает достаточно времени, так зачем тратить часы на копирование вашего контента на другие платформы? Чем меньше времени вы тратите на утомительную работу, тем больше времени вы можете потратить на то, что действительно важно: на свой контент.
В конечном счете, вы можете больше говорить, делать больше и охватывать большее количество участников без чрезмерного использования внутренних ресурсов. (Victoria Fox, Blackboard Blog)P ublish multi-channel with StoryChief
Еще одна важная вещь, которую вы всегда должны помнить о добавлении, — это призыв к действию и / или форма захвата лида. Вы хотите собрать информацию о людях, проявивших интерес к вашей статье, и сохранить их в своей базе данных! Расширяйте свою аудиторию и регулярно присылайте им новые обновления.
В StoryChief вы можете легко встроить форму захвата лидов, независимо от того, подключена ли она к вашей CRM-системе, списку MailChimp или просто для получения данных прямо на вашу электронную почту. Вот пример того, как это выглядит:
Этот список ни в коем случае не является исчерпывающим. Есть много других способов улучшить ваше длинное чтение, например, иметь интригующий дизайн, добавить инфографику, включая достаточное количество примеров, использовать элементы повествования и т. Д. Позвольте своему творчеству направлять вас, и вы увидите, как ваша контент-стратегия значительно улучшится.
Не знаете с чего начать? Вот несколько полезных руководств, которые мы создали для вас:
Улучшите написание контента и ознакомьтесь с нашими советами по оптимизации вашей контент-стратегии!
Клуб долгого чтения | Клуб долгого чтения
Клуб долгого чтения является частью недавно финансируемой премии Wellcome Trust Technology Development Award. Наша общая цель — помочь сообществу специалистов по геномике достичь готового качества, полных геномов эталонного качества на регулярной основе и в соответствующем масштабе.
Список рассылки
Первое правило клуба long read — подпишитесь на уведомления по электронной почте, чтобы узнавать о предстоящих онлайн-событиях !.
Канал на YouTube
Наш канал на YouTube находится по адресу:
https://www.youtube.com/c/longreadclub
Эпизод 1: Что такое клуб долгого чтения?
Эпизод 2: Извлечение ДНК с Джилл Хершлеб, 10X Genomics
Эпизод 3: Сборки полного генома человека с Адамом Филлиппи, NHGRI
Эпизод 4: Сборщик Flye с Михаилом Колмогоровым и Джеффри Юаном, UCSD
Эпизод 5: Представляем MorphoSeq: высокоточные длинные считывания с платформ короткого считывания, Аарон Дарлинг, Longas Technologies
Протоколы.io
Зайдите в нашу группу Protocols.io по адресу:
https://www.protocols.io/groups/longreadclub
Наборы данных
Эта страница содержит ссылки на образцы или известные наборы данных для длительного чтения:
Nanopore NA12878 Консорциум
Консорциум нанопор человека WGS курирует регулярно обновляемую линию клеток человека NA12878 освобождение данных от данных нанопор. Больше информации на их Github:
https://github.com/nanopore-wgs-consortium/NA12878
Консорциум теломер-теломеры
На AGBT 2019 консорциум T2T представил обширный набор данных для гаплоидной клетки человека CHM13. включает в себя 50-кратное сверхдлинное покрытие нанопор, 10-кратное увеличение данных геномики, данные BioNano.Это дополняется существующими данными PacBio. Цель консорциума — завершить построение теломер-теломеры. непрерывная завершенная сборка гаплоидного генома человека.
Узнайте больше на их веб-сайте: https://sites.google.com/ucsc.edu/t2tworkinggroup/home Выпуск данных доступен через GitHub: https://github.com/nanopore-wgs-consortium/CHM13
Набор данных сверхдлительного чтения E. coli
В рамках разработки протокола мы сгенерировали обширные сверхдлинные данные чтения для Э.coli K-12 MG1655, который выпущен здесь на случай, если он будет полезен для разработки (например, программного обеспечения для сборки).
Длинное секвенирование выявляет структурные вариации генома, лежащие в основе создания качественного белка кукурузы
Сборка и проверка генома
Геном K0326Y был секвенирован и собран с использованием трех технологий: одномолекулярное секвенирование PacBio в реальном времени (SMRT), парное соединение Illumina -концевое секвенирование и оптическое картирование BioNano (методы). Исходный геном K0326Y был собран в 2148 Mb с использованием 28.35 миллионов длинных чтений с длиной чтения N50 в 16,6 кб и примерно 139-кратным покрытием (дополнительные таблицы 1–3 и дополнительный рисунок 1), в результате получается 1221 контиг с N50 в 6,99 МБ (таблица 1, дополнительные таблицы 4, 5). ), что сопоставимо с недавно опубликованным геномом SK с контигом N50 5,93 Mb (ref. 9) . Собранные контиги были скорректированы с помощью согласованных последовательностей PacBio 132,5 ГБ и высококачественных считываний парных концов Illumina на 217,5 ГБ. Эти обработанные контиги были подвергнуты гибридной сборке с помощью оптических карт, созданных из 389.3 Гб молекул BioNano. Сборка содержала 870 каркасов с размером каркаса N50 27,98 МБ (таблица 1 и дополнительная таблица 5). Эталонный геном B73 10 использовали для картирования и ориентации каркасов на хромосомах K0326Y, на которые приходилось 97,74% (2112 Mb) от всех собранных последовательностей (дополнительная таблица 6). Общий размер сборки генома K0326Y составлял 2161 МБ, аналогично недавно опубликованным B73 (2106 МБ) 10 и Mo17 (2183 МБ) 11 . Однако у K0326Y было только 438 пропусков и окончательный размер контига N50 равнялся 7.77 Mb, обеспечивая в 5 раз более высокие смежные последовательности, чем B73 (contig N50: 1,25 Mb) и Mo17 (contig N50: 1,47 Mb) (таблица 1 и дополнительный рисунок 2).
Таблица 1 Глобальная статистика сборки генома K0326Y.Для оценки качества собранного генома K0326Y была использована генетическая карта пангенома кукурузы с высокой плотностью, содержащая ~ 4,4 миллиона тегов «генотип путем секвенирования» (GBS) 12 . Выравнивание генома K0326Y с заякоренными тегами GBS показало высокую согласованность в отношении положения и ориентации картированных каркасов (дополнительный рис.3). Точность и полнота сборки подтверждены 100-кратным считыванием данных Illumina с коэффициентом картирования 93,8% (дополнительная таблица 2). Приблизительно 95,8% (1380 из 1440 генов) генов эмбриофитов были обнаружены в сборке K0326Y согласно BUSCO 13 , процент аналогичен таковому для генома B73 (96,1%) и Mo17 (95,4%) (дополнительная таблица 7).
Анализ повторений
Непрерывные последовательности представляют собой серьезное улучшение для хромосомных областей с высоким содержанием повторяющихся последовательностей 10 (рис.2). В общей сложности 83,32% генома K0326Y состояло из повторяющихся элементов, включая ретротранспозоны (77,38%), ДНК-транспозоны (4,72%) и неклассифицированные элементы (0,49%) (дополнительная таблица 8). Из ретротранспозонов в 10 хромосомах было идентифицировано 136 191 интактных длинных концевых повтора (LTR) с высокой степенью достоверности, что немного больше, чем сообщалось для B73 (ref. 10 ). Семейства ретротранспозонов Gypsy и Copia составляли ~ 43,44% и 23,74% соответственно собранных последовательностей K0326Y.Общий состав семейств ретротранспозонов в K0326Y был очень похож на таковой в геномах Mo17 и B73, поскольку они имели место до одомашнивания (дополнительный рис. 4). Тем не менее, несколько семейств показали вариации числа копий среди K0326Y, B73 и Mo17, например, семейство LTR Ty1 / Copia RLC_ebel , с пятью копиями в B73, но 159 и 154 копиями в K0326Y и Mo17. Было 647 копий Ty3 / Gypsy RLG_huck_ AC214833 в B73, по сравнению с 482 и 446 копиями в K0326Y и Mo17, соответственно.Более похожий образец числа копий семейства LTR наблюдался у K03236Y и Mo17 по сравнению с K03236Y и B73. Центромерные области для каждой хромосомы были реконструированы с помощью анализа связанных с центромерой длинных концевых повторов (CRM) (дополнительная таблица 9) и тандемных повторов длиной 156 п.н. (CentC) 14 (дополнительная таблица 10), демонстрируя аналогичный образец распределения, как у B73. (Дополнительные рисунки 5, 6).
Рис. 2: Геномный ландшафт генома K0326Y.a Плотность переносных элементов. b генная плотность. c Уровни экспрессии генов. d СНП. e Indels. f Распределение PAV. Для дорожек d , e и f внешний слой — B73, а внутренний — Mo17. Скользящее окно для всех треков составляет 1 Мб. Исходные данные представлены в виде файла исходных данных.
Аннотации генома
Всего 1618691 высококачественное полноразмерное нехимерное считывание (HQ-FLNC), т. Е. Полноразмерные консенсусные последовательности кДНК, было идентифицировано в результате секвенирования изоформы PacBio, что дало 247616 неизбыточных транскриптов для прогнозирования. генные модели с использованием маркера-P 10,15 (дополнительная таблица 11).Набор из 38 238 генов с 60 475 транскриптами был идентифицирован в геноме K0326Y (дополнительная таблица 11), что сопоставимо с B73 (39 200 генов) 10 и Mo17 (38 620 генов) 11 . Данные полноразмерной кДНК улучшили аннотацию генома K0326Y за счет того, что 69% генных моделей поддерживались полноразмерными транскриптами (охват CDS> 50%) (дополнительная таблица 12). Кроме того, 37 861 (99,01%) генная модель K0326Y может быть отнесена к определенным хромосомным участкам.
Сравнительная геномика
Понимание внутривидовой изменчивости кукурузы имеет важное значение для улучшения сельскохозяйственных культур и селекции растений.Учитывая смежные последовательности генома, мы смогли исследовать структурное разнообразие между тропическим K0326Y и двумя инбредными животными умеренного климата, B73 (ref. 10 ) и Mo17 (ref. 11) . Приблизительно 58% последовательностей генома K0326Y совпадают один к одному синтеническим блокам B73 и Mo17, соответственно (дополнительная фиг. 6). Пропорция синтенных областей между K0326Y и B73 или Mo17 по всему геному была ниже, чем недавний анализ между B73 и Mo17 (ref. 11 ), возможно, из-за усиленного дивергентного отбора урожая семян и репродуктивного успеха между B73 и Mo17, что важно для гетерозиса между двумя гетеротическими группами 16 .Было обнаружено две инверсии, характерные для K0326Y по сравнению с линиями кукурузы умеренного и тропического климата. Хромосома 1 содержала одну большую перицентрическую инверсию размером 8,5 Мб, которая была подтверждена оптической картой BioNano с их точками разрыва (дополнительные рисунки 6, 7). Инверсия была другой: одна из 1,7-Mb SK хромосомы 1 произошла из Южной Америки, что указывает на то, что эта структурная вариация может быть специфичной для K0326Y 9 . Сообщалось, что некоторые гены в инвертированной области проявляли значительную связь со временем цветения кукурузы 17 .Другая большая парацентрическая инверсия размером 5,8 млн п.н. была расположена в центромере хромосомы 4 (дополнительные рисунки 6, 7). Большинство генов в инверсии имеют функции, связанные с углеводным обменом и регуляцией генов. K0326Y представляет собой линию QPM, несущую мутацию o2 . Сравнение гена O2 в K0326Y с B73 и Mo17 показало, что имелась вставка транспозона rbg длиной 4958 п.о. на 249 п.н. перед стартовым кодоном (ATG), аналогично другим аллелям o2 (дополнительный рис.8) 18 . Как и ожидалось, данные по экспрессии генов показали, что транскрипция O2 ингибируется из-за вставки rbg .
Геномный полиморфизм и структурные вариации
Геном кукурузы демонстрирует очень высокий уровень генетического разнообразия в отношении SNP, малых InDel и структурных вариаций, которые вносят вклад в фенотипическое разнообразие и гетерозис у гибридов кукурузы 19 . Мы идентифицировали в общей сложности 10 205 511 SNP и 1 397 901 InDel (<100 п.н.) между K0326Y и B73, в среднем 8.35 SNP и 1,14 InDels на килобазу. Между K0326Y и Mo17 было 9655364 SNP и 1 458 329 InDel (<100 пар оснований), в среднем 7,77 SNP и 1,17 InDel на килобазу (дополнительная таблица 13). Генетический полиморфизм затрагивает 8702 гена в B73 и 6009 генов в Mo17, включая сдвиг рамки считывания, потерю стоп-кодонов и приобретение стоп-кодонов (дополнительный рис. 9), что может способствовать другим функциям в K0326Y. Мы идентифицировали 19 778 вставок (> 100 п.н.) и 39 931 делецию между K0326Y и B73, которые могли затронуть 6 538 и 10 463 генов, соответственно.В случае K0326Y и Mo17 32 071 вставка могла повлиять на 9 323 гена, а 46 461 делеция была обнаружена в 12 456 генах, соответственно (дополнительные таблицы 14, 15).
Было идентифицировано распределение вариаций присутствия / отсутствия (PAV), которые присутствовали только в K0326Y, но полностью отсутствовали в B73 и Mo17. Всего в K0326Y было идентифицировано 39 479 сегментов, при этом общая длина 154,7 Мбайт отсутствовала в B73. Точно так же в K0326Y было 37906 сегментов с общей длиной 149,5 Мб, отсутствующих в Mo17 (рис.2). Эти области PAV затронули 3568 генов в K0326Y (дополнительные данные 1). Экспрессия 631 гена PAV была повышена на основании сравнения данных РНК-seq из развивающегося эндосперма 16-DAP K0326Y QPM по сравнению с W64A o2 . Эти гены были обогащены такими путями, как биосинтез крахмала и метаболические процессы, активность АТФазы, биосинтез ауксина и трансмембранная активность серы (дополнительный рисунок 10). Интересно, что было обнаружено, что гены PAV в B73, Mo17 и Ph307 также присутствуют у диких родственников кукурузы и встречаются до завершения одомашнивания кукурузы, но после расхождения сорго и кукурузы 11 .
Дупликации генов обеспечивают механизм изменения фенотипов посредством эффекта дозировки гена или функции гена от дивергенции. Мы обнаружили, что количество одноэлементных генов в B73, Mo17 и K0326Y было вполне консервативным, тогда как K0326Y имел меньше сегментарных дупликаций (12 259), но более рассредоточенные дупликации генов (16 777) по сравнению с B73 и Mo17 (дополнительная таблица 16). Тандемно дублированные гены были расположены в ближайших окрестностях и потенциально имели одни и те же регуляторные элементы. Мы идентифицировали 1261 тандемный кластер копий гена, что составляет 3842 аннотированных гена в геноме K0326Y (дополнительная таблица 16).
Генетическое картирование
Mo2 sУчитывая повсеместную генетическую изменчивость между QPM и другими мутантами o2 , ожидалось, что определенные вариации, включая гены Mo2 , ответственны за их фенотипические различия. Чтобы картировать хромосомные области, связанные с модификацией эндосперма, мы следовали методам Holding 3,4 и скрестили K0326Y с W64A o2 . Объединенную ДНК из ткани листа F 2 стекловидного тела и непрозрачных ядер использовали для анализа объемной сегрегации с секвенированием следующего поколения (BSA-seq) для идентификации QTL, связанных с модификацией эндосперма.Чтобы избежать маскирующего эффекта со стороны основного Mo2 ( qγ27 ), мы выбрали стекловидное тело и непрозрачные ядра с двумя копиями гена γ27 в локусе γ27 для анализа QTL-seq 8 (дополнительный рисунок 11). ). Три Mo2 QTL были расположены на хромосомах 1, 7 и 9 (рис. 3b и дополнительный рис. 12), что согласуется с данными предыдущего картирования 3,4 . Другой QTL с острым пиком был обнаружен в области на хромосоме 6 с большой вставкой K0326Y-специфического фрагмента (дополнительный рис.3). Независимо от того, является ли это ложноположительным результатом, необходимо дополнительное исследование, поскольку кроссоверы в популяции F 2 очень ограничены.
Рис. 3: Отображение модификаторов o2 .a Построение популяции F 2 с использованием K0326Y (QPM) и W64A o2 ( o2 ). K0326Y имеет фенотип стекловидного тела, а W64A o2 имеет непрозрачный фенотип. b картирование модификаторов o2 с помощью анализа объемной сегрегантности (BSA) сегрегационной популяции, полученной в результате скрещивания K0326Y и W64A o2 .Значение G ’представляет собой сглаженную версию стандартной статистики G в каждом скользящем окне размером 4 Мбайт. Зеленая линия указывает порог значения G ’, соответствующий FDR 8 × 10 -7 . Масштабная линейка = 5 мм.
Для идентификации генов-кандидатов, соответствующих Mo2 QTL, суммарную РНК из развивающегося эндосперма кукурузы собирали на 16 DAP из стекловидного тела и непрозрачных ядер. Транскриптомные профили были построены для сравнения уровней экспрессии генов между K0326Y и W64A o2 и между CM105 Mo2 (другая линия QPM на фоне CM105) и CM105 o2 .Было 1791 DEG, которые перекрывались в двух сравнениях, из которых 926 были активированы, а 865 — подавлены (дополнительные данные 2). Увеличенные ДЭГ были значительно обогащены с точки зрения связывания шаперона, связывания развернутого белка, сворачивания белка и реакции на нагревание и температурный стимул (дополнительный рис. 13). Примечательно, что 43 гена, кодирующие белок теплового шока (HSP) и факторы транскрипции HSP, были значительно активированы в QPM (дополнительные данные 3). Как предполагалось ранее 20 , они могут активироваться и уменьшать стрессовые эффекты путем повторной сборки развернутых или агрегированных белков, созданных мутацией o2 .
Было обнаружено 216 (117 с усиленной регуляцией и 99 с пониженной регуляцией) DEG, которые перекрестно проверялись с генами из анализа QTL-seq, сужая пул генов-кандидатов, вносящих вклад в фенотип стекловидного тела и адаптацию к окружающей среде (дополнительные данные 4). После сортировки списка 216 генов было 125, содержащих SNP, 44 с инсерциями (> 100 п.н.) и 43 с делециями (> 100 п.н.) по сравнению с B73, а также 18 с тандемными дупликациями и 19 принадлежащих PAV ( Дополнительные данные 4).Тем не менее, большинство DEGs, которые не могут быть картированы в QTL, необязательно могут быть причиной фенотипа QPM, но они могут дать ключ к разгадке влияния Mo2s, которые запускают сигнальный каскад, ведущий к образованию эндосперма стекловидного тела. Они также могут находиться под регуляцией Mo2s, отражая последующие эффекты изменений в составе ядра.
Когда мы интегрировали вариации генома для идентификации генов-кандидатов, связанных с Mo2 s, которые заслуживают дальнейшего изучения, стали очевидными те, которые имеют аллельные вариации, повышенную экспрессию и расположены в областях QTL.На хромосоме 1 было удалено 85 п.н. из промотора O10 в K0326Y, который был высоко экспрессирован в линиях K0326Y и CM105 Mo2 QPM. O10 кодирует специфический для злаков белок, который регулирует отложение и организацию зеина в PB 21 . Мутации в O10 создают аномальное распределение зеинов в PB. Остается исследовать, действует ли повышающая регуляция O10 как Mo2 или необходима для опосредованной Mo2 модификации эндосперма.
Тандемная дупликация локуса γ27 ( qγ27 ), который ранее был обозначен как аллель Standard ( S ) для несения двух копий гена γ27 , является основным Mo2 для модификации эндосперма в КПМ 8 . Помимо γ27 , эта дупликация включает три других гена (GRMZM2G565441, GRMZM2G138976 и GRMZM5G873335) на основе B73_vs3, которые были объединены в один ген с пятнадцатью экзонами, кодирующими фактор транскрипции ARID 4 (Zm00001d0205
) .Хотя все линии QPM имеют эту дупликацию, непрерывная геномная последовательность ранее не была доступна, и ее вариация была неизвестна 8 . Здесь мы смогли расширить эту последовательность до 28 т.п.н., полностью покрывая дупликацию (~ 15 т.п.н.). Дублированные фрагменты имеют множество SNP, которые могут их отличить. Первая копия ARID4 имеет делецию 1923 п.н. на своем 3 ’конце, что приводит к отсутствию четырех экзонов по сравнению со второй копией гена (дополнительный рисунок 14). Пара полиморфных праймеров для ПЦР, фланкирующая эту делецию, может быть разработана для выбора дупликации во время разведения QPM в зависимости от наличия двух полос ПЦР.Глобальное выравнивание показало, что локус qγ27 между Mo17 и K0326Y был почти идентичен. Аллель B73- γ27 был ближе к первой копии фрагмента в K0326Y, а B73- ARID4 был ближе ко второй копии в K0326Y (дополнительный рис. 15). Это указывает на то, что однокопийный аллель γ27 в B73 мог быть получен в результате реаранжировки ДНК дублированного аллеля. Следовательно, современные линии кукурузы могли иметь по крайней мере три типа однокопийного аллеля γ27 , тогда как тандемная дупликация могла произойти раньше, чем одомашнивание кукурузы (дополнительный рис.16).
В соответствии с предыдущими исследованиями, наш анализ картирования QTL (BSA-seq) и экспрессии генов (RNA-seq) идентифицировал другой многообещающий ген-кандидат Mo2 на хромосоме 9, Pfpα 3,4 . Pfpα кодирует α-регуляторную субъединицу пирофосфат-зависимой фруктозо-6-фосфат-1-фосфотрансферазы и служит не требующим АТФ ферментом во время гликолиза. Уровни транскрипта и белка, а также активность фермента Pfpα были значительно увеличены в эндоспермах QPM по сравнению с нормальными и мутантами o2 3,4,20 .Как отмечалось, это может приводить к увеличению гликолитического потока в эндосперме QPM и улучшать аспект фенотипа o2 , включая ограничение в ATP 20 . Идентичность причинных полиморфизмов и лежащих в основе механизмов экспрессии, влияющих на Pfpα , не была выяснена из-за отсутствия последовательности генома QPM. Мы обнаружили, что локус Pfpα в K0326Y обнаруживает драматические структурные вариации по сравнению с Mo17 и B73. В K0326Y -Pfpα имелся 983-п.н. Helitron в промоторе и вставка из 2485-п.н. во втором интроне, по сравнению с Mo17- Pfpα и B73 -Pfpα .Однако Mo17- Pfpα и B73 -Pfpα оба содержали CACTA длиной 6181 п.о., ретротранспозон длиной 10,685 п.о. во втором интроне и ретротранспозон длиной 6037 п.н. в 13-м интроне по сравнению с K0326Y -Pfpα. (рис. 4а). Чтобы исследовать, влияют ли эти вставки и делеции на экспрессию Pfpα , транскрипты каждого экзона были нормализованы по размеру экзона и глубине секвенирования. Мы обнаружили, что количество транскриптов всего гена и каждого экзона Pfpα в QPM (K0326Y и CM105 Mo2 ) было выше, чем в линиях без QPM (W64A o2 , CM105 o2 , CM105 +) (рис.4b и дополнительный рис. 17). Кроме того, мы обнаружили, что 65% линий QPM имеют вставку Helitron . Вставка Helitron сохранялась в 95% ядер эндосперма стекловидного тела сегрегирующей популяции F 2 (рис. 4c) и, следовательно, четко ассоциирована с фенотипом ядра стекловидного тела. Мобильные генетические элементы могут управлять эволюцией генома, а также изменять экспрессию генов путем встраивания в интроны, экзоны или регуляторную область. Опосредованные транспозоном мутации в промоторе Adh2 привели к значительному увеличению экспрессии гена в пыльце 22 , и подействовал мобильный элемент, вставленный в регуляторную область гена одомашнивания кукурузы ( teosinte branch1 , tb1 ). как усилитель его экспрессии, что частично объясняет усиление апикального доминирования у кукурузы 23 .Вставка Helitron в промотор Pfpα может быть частично причиной модификации эндосперма в QPM. Влияние других мобильных элементов на функциональные вариации требует дальнейшего исследования.
Рис. 4: Pfpα , связанный с модификацией эндосперма o2 .a Структурная вариация Pfpα среди K0326Y, Mo17 и B73. Структура гена Pfpα показана с экзонами в синих прямоугольниках и интронами с черной линией. Helitron в промоторе помечен зеленой полосой. Вставки ретротранспозона LTR, сокращенно LTR, показаны оранжевыми перевернутыми треугольниками. Остальные вставки представлены фиолетовыми перевернутыми треугольниками. Физическое местоположение основано на геноме K0326Y. ( b ) Относительная экспрессия гена Pfpα для дикого типа (CM105 +), o2 (CM105 o2 и W64A o2 ) и QPM (CM105M o2 и K0326Y). P -значения определяются двусторонним тестом Стьюдента t .Планки погрешностей определяются как стандартное отклонение выборки от размера выборки (биологически независимые выборки) n = 5. c Распределение вставок Helitron в инбредных линиях QPM, популяции GWAS и K0326Y X W64A o2 F 2 сегрегация населения; Ось x показывает популяции кукурузы, а ось y представляет собой связанный процент. Исходные данные, лежащие в основе рис. 4b, c представлены в виде файла исходных данных.
Среди кандидатов Mo2 два гена, SR45a и ERDJ3A , были чрезвычайно близки к пику на хромосоме 9 (дополнительные данные 4).Уровень транскриптов SR45a увеличивался в 2–28 раз в QPM по сравнению с мутантами o2 (дополнительный рисунок 18 и дополнительные данные 4). Этот ген отвечал за сплайсинг РНК, и его уровень также повышался, когда Arabidopsis находился в состоянии стресса 24 . Интересно, что в 5-м интроне SR45a был транспозон ДНК из 399 п.н. элемента hAT по сравнению с B73 и Mo17 и в этой области с соответствующим индексом 61% (дополнительный рис.18), был тесно связан с признаком стекловидного тела в популяции F 2 . Сообщалось, что ERDJ3A является ко-шапероном с белками теплового шока (HSP), экспрессируемыми в растительных клетках. Ген может в значительной степени индуцироваться при стрессе ER у риса и способствует доставке развернутых белков между вакуолями и ER 25 . Мы обнаружили, что его экспрессия была увеличена в 5-8 раз в QPM по сравнению с o2 , и что ретротранспозон длиной 26 022 п.н. был расположен ниже последовательности ДНК ERDJ3A (дополнительный рис.19), которые могут регулировать экспрессию гена из-за его большого размера и пространственного изменения. Влияет ли вставка на экспрессию генов и, в свою очередь, играет роль в модификации эндосперма, еще предстоит изучить.
Секвенирование длинных транскриптомов показывает большое разнообразие промоторов в различных молекулярных подтипах рака желудка | Genome Biology
Пейзаж длинночитаемых полноразмерных изоформ в клеточных линиях GC
Чтобы получить репрезентативный обзор полноразмерных транскриптов в GC, мы выполнили долгосрочное секвенирование РНК PacBio на десяти линиях клеток GC.Линии GC были отобраны для представления четырех подтипов TCGA GC (CIN — хромосомно нестабильный, EBV — вирус Эпштейна-Барра — положительный, GS — геном стабильный и MSI — микросателлитный нестабильный) на основе предшествующей литературы и внутреннего молекулярного анализа [5] ( Дополнительный файл 2, таблица S1). Для каждой строки мы сгенерировали ~ 26 ГБ необработанных данных секвенирования и использовали модуль циклической согласованной последовательности программы IsoSeq3 (https://github.com/PacificBiosciences/IsoSeq3) для генерации согласованных чтений (рис. 1a). Консенсусные чтения были отфильтрованы для полноразмерных нехимерных (FLNC) чтений.Чтобы идентифицировать уникальные изоформы, чтения FLNC были подвергнуты кластеризации de novo с использованием модуля IsoSeq3cluster. Все изоформы были картированы в геном человека (версия hg38) с использованием GMAP [14], и только высококачественные изоформы (поддерживаемые как минимум двумя чтениями FLNC) рассматривались для дальнейшего анализа. В среднем мы идентифицировали ~ 37700 неизбыточных полноразмерных изоформ на клеточную линию. Дальнейший контроль качества и аннотации изоформ были выполнены с использованием SQANTI2 (https://github.com/Magdoll/SQANTI2), что дало в среднем ~ 27000 аннотированных изоформ на строку.Эти результаты сопоставимы с ранее опубликованными данными Iso-seq клеточной линии человека [12, 15].
Рис. 1Пейзаж длинночитываемого транскриптома в клеточных линиях рака желудка. a Алгоритм вызова изоформы для данных Iso-seq. b Типы и иллюстрации идентифицированных изоформ. c Распад идентифицированных изоформ. d Метрика контроля качества для предсказанных изоформ. По сравнению с расшифровками FSM с высокой степенью достоверности, NIC и NNC имеют схожую метрику качества.ISM имеет низкий процент транскриптов с поддержкой CAGE, что позволяет предположить, что некоторые из этих транскриптов происходят из-за 5′-деградации. e Количество изоформ на ген. f Количество клеточных линий на изоформы. г Число обнаруженных изоформ по сравнению с числом профилированных клеточных линий. Известные транскрипты FSM (красный) достигают насыщения, в то время как новые транскрипты постоянно обнаруживаются
В общей сложности мы идентифицировали 60 239 неизбыточных изоформ транскриптов в десяти линиях GC и классифицировали изоформы на четыре группы на основе базы данных эталонных транскриптомов человека Gencode v32. (Рисунок.1b представляет собой иллюстративную карикатуру). Среди изоформ 31% (18442) соответствовали полному сплайсингу (FSM), полностью совпадающему с известными транскриптами, а 37% (21874), 29% (17333) и 3% (1709) были новыми в каталоге (NIC; соответствующие к изоформам, по крайней мере, с одним неаннотированным сайтом сплайсинга), новым, не указанным в каталоге (NNC; соответствует изоформам с известными сайтами сплайсинга, но новыми соединениями сплайсинга) и неполным совпадениям сплайсинга (ISM; соответствует изоформам, которые соответствуют части известного расшифровка) (рис.1в). Мы использовали различные функции качества, предоставляемые SQANTI2, для оценки надежности полноразмерных изоформ, включая использование неканонических соединений, внутренние свойства секвенирования (т. Е. Количество предсказанных артефактов переключения шаблона обратной транскриптазы) и функциональные геномные доказательства, такие как перекрытие изоформ. 5′-транскрипт заканчивается независимо опубликованными данными Cap Analysis of Gene Expression (CAGE) [16] (CAGE включает данные секвенирования метки, непосредственно измеряющие 5′-конец транскрипта), а 3′-концы — хвостами полиА (рис.1г). Сопоставляя новые изоформы с известными изоформами высокого качества (FSM), мы обнаружили, что новые изоформы NIC и NNC демонстрируют качество, сопоставимое с известными изоформами, в то время как ISM демонстрируют меньшую долю перекрытия с пиками CAGE. Возможно, что некоторые изоформы ISM могут содержать частичные фрагменты, возникающие в результате неполной ретротранскрипции или артефактов распада мРНК [17]. В связи с этим мы исключили изоформы ISM (3%) из последующего анализа.
Помимо категорий изоформ FSM, NNC, NIC и ISM, SQANTI2 также генерирует небольшое количество транскриптов, классифицируемых как антисмысловые ( n = 261; 0.4%), генные ( n = 304; 0,5%; изоформы, которые перекрываются с интроном) и межгенные ( n = 316; 0,5%; изоформа в межгенных областях) (см. Дополнительный файл 2; таблица S2). Предыдущие исследования показали, что эти изоформы имеют тенденцию быть одноэкзонными с более высоким процентом неканонических сплайсинговых соединений, что может быть вызвано экспериментальными или техническими артефактами [17]. По этим причинам и небольшому количеству изоформ в этих категориях (<1%) мы не рассматривали эти категории (антисмысловые, генные, межгенные), и наше исследование сосредоточено только на альтернативных событиях сплайсинга, обнаруженных в FSM, NIC и NNC. категории ( n = 57 649).
Изоформы транскрипта ( n = 57 649) картированы более чем с 14 K генами ( n = 14 203), при этом 67% генов связаны с более чем 1 изоформой (9462 гена). Каждый ген был ассоциирован в среднем с 2 изоформами (рис. 1e). Большинство изоформ (33 271, 58%) экспрессировалось только в одной клеточной линии, а 3513 изоформ (6%) были обнаружены во всех клеточных линиях (рис. 1f). Интересно, что анализ кривой разрежения показал, что, хотя при обнаружении известных изоформ наблюдается насыщение, открытие новых изоформ остается ненасыщенным (рис.1г). Чтобы оценить обнаружение изоформ в зависимости от глубины секвенирования, мы также выполнили анализ разрежения в каждой отдельной клеточной линии путем подвыборки количества чтений полной длины. Как показано в Дополнительном файле 1; На рисунке S1 мы обнаружили, что для каждой клеточной линии при глубине секвенирования 26 Гб обнаружение изоформ достигло насыщения. Таким образом, увеличение количества новых изоформ в клеточных линиях более вероятно связано с транскриптомами, специфичными для клеточных линий, а не с отсутствием покрытия в отдельных клеточных линиях.Эти анализы показывают, что изучение транскриптомического ландшафта новых изоформ остается богатой областью неиспользованного биологического разнообразия.
Чтобы изучить взаимосвязь между альтернативными событиями сплайсинга и соматическими изменениями, мы объединили данные альтернативного сплайсинга с соматическими мутациями, идентифицированными путем секвенирования всего экзома 10 линий. Соматические мутации были идентифицированы с использованием Mutect2 [18] в режиме «только опухоль» с вычитанием зародышевой линии с использованием базы данных gnomAD [19] и панели из 36 нормальных образцов экзома.Соматические изменения были дополнительно аннотированы с помощью Funcotator, и все варианты, классифицированные как Splice Site, были проверены. Этот анализ выявил в общей сложности 335 мутаций сайтов сплайсинга в 10 линиях. Ручная проверка этих мутаций в IGV выделила 49 из этих мутаций, которые могут привести к изменениям в сплайсинге мутировавших экзонов, как обнаружено по данным Iso-seq. Это указывает на то, что подавляющее большинство идентифицированных изменений сплайсинга (изоформы NIC и NNC; n = 39,207 по сравнению с 49) вызваны дерегуляцией транскрипции, а не соматическими изменениями.
Характеристики долго считываемых новых изоформ
Мы приступили к характеристике новых изоформ транскриптов. Из 39 207 новых изоформ 17 333 (44%) были классифицированы как NNC, а остальные новые изоформы были NIC. Мы наблюдали изоформы NNC и NIC с участием связанных с раком генов, таких как ERBB2 и CD44 , и подтвердили ранее известные изоформы FSM, связанные с этими двумя генами (рис. 2a). Например, мы идентифицировали изоформу NNC ERBB2 с альтернативным сайтом сплайсинга 3′-экзона в экзоне 26.Предполагается, что это событие сплайсинга вызовет потерю 14 аминокислот, частично удаляя домен тирозинкиназы ERBB2 . В качестве другого примера мы идентифицировали изоформу NIC CD44 , напоминающую известную вариантную изоформу CD44-209, но с дополнительным экзоном 6a, что привело к усилению домена, подобного Herpes_BLLF1 (CD-поиск, e-value = 2,3 × 10 — 3 ).
Рис. 2Характеристики транскриптов Iso-seq. a Примеры событий альтернативного сплайсинга ( CD44, ) и альтернативного промотора ( ERBB2 ), идентифицированных в данных Iso-seq.Порядок экзонов обозначен внизу, а перекрывающиеся экзоны обозначены алфавитами. Для ERBB2 помечены только перекрывающиеся экзоны. Новые экзоны (относительно аннотации Gencode) были указаны черным. Аннотации белкового домена для выбранных изоформ были показаны на нижней панели. b Характеристики новых (NIC и NNC) и известных (FSM) транскриптов. NIC и NNC имеют больше экзонов, более длинные изоформы и CDS. Новые транскрипты могут генерировать новые TSS и TTS. c Новые изоформы с большей вероятностью будут мишенями для NMD, экспрессируются на более низком уровне и содержат больше сайтов связывания MHC, чем известные изоформы
По сравнению с известными изоформами, новые изоформы транскриптов (как NNC, так и NIC) обладают большим количеством экзонов (медиана 13 против 8, тест Вилкоксона, p значение <2.2 × 10 — 16 ), более длинные транскрипты (медиана 3593 против 2986,5 п.н., критерий Вилкоксона, значение p <2,2 × 10 — 16 ) и последовательности, кодирующие белок (медиана 1593 по сравнению с 1260 п.н., критерий Вилкоксона, p значение <2,2 × 10 — 16 ) (рис. 2б). Новые изоформы также с большей вероятностью приобретали новые сайты начала транскрипции (TSS) (11% изоформ более чем на 1 kb от известных TSS против 1%; тест Фишера p значение <2,2 × 10 -16 ) и сайты терминации (TTSs). ) (14% изоформ на расстоянии более 1 т.п.н. от известных TTS против 8%; тест Фишера p значение <2.2 × 10 — 16 ). Использование новых сайтов терминации также было связано с более высокой вероятностью преждевременных стоп-кодонов, связанных с нонсенс-опосредованным распадом мРНК (NMD) (22% против 7%; тест Фишера p значение <2,2 × 10 — 16 ). Затем мы использовали Kallisto [20], чтобы вывести уровни экспрессии полноразмерной изоформы из данных короткого считывания РНК-seq и netMHCpan [21], чтобы идентифицировать потенциальные антигенные пептиды из аннотированных изоформ. Новые изоформы также экспрессировались на более низких уровнях по сравнению с известными изоформами (медиана TPM 0.54 против 3,08; Тест Вилкоксона, p , значение <2,2 × 10 — 16 ) и содержал более высокую долю сайтов связывания главного комплекса гистосовместимости (MHC) (медиана 5,6 сайта связывания на kb против 5,4; тест Вилкоксона, p значение 9,8 × 10 -7 ) (рис. 2в). Необходимы дальнейшие исследования, чтобы определить, связаны ли эти предсказанные различия аффинности связывания MHC с биологически релевантными паттернами иммуногенности и Т-клеточными ответами.
Затем мы сравнили уровни транскриптома и профиля экспрессии, полученные с использованием методов длинного чтения Iso-seq или короткого чтения RNA-seq.Мы выполнили открытие изоформ на коротко читаемой РНК-seq Illumina с использованием Stringtie (на основе ссылок и с учетом новых изоформ) в тех же десяти линиях клеток, профилированных Iso-seq. Подобно анализу Iso-seq, идентифицированные изоформы были аннотированы с помощью SQANTI2 и сгруппированы в соответствии с категориями FSM, NIC и NNC. Используя gffcompare, мы сравнили методы длительного чтения и короткого чтения и обнаружили, что большинство изоформ, идентифицированных в результате длительного чтения (66,8%), не могут быть легко восстановлены только из данных короткого чтения (дополнительный файл 1; рисунок S2A).Более того, хотя анализ короткого чтения только для короткого чтения выявил большее количество изоформ, дальнейшее исследование показало, что по сравнению с анализом длительного чтения данные короткого чтения имели гораздо более короткие длины изоформ (2182 п.н. против 3564 п.н., t -тест p значение <2,2 × 10 — 16 ), содержал меньше экзонов (7,2 против 13,1, t -тест p значение <2,2 × 10 — 16 ) и менее сильно поддерживался CAGE (38,3 % против 70,3%, тест Фишера p значение <2.2 × 10 — 16 ) и данных polyA (45,0% против 81,3%, тест Фишера p значение <2,2 × 10 — 16 ). Эти результаты предполагают, что многие предсказанные изоформы короткого чтения являются неполными фрагментами полноразмерных изоформ, а не истинными изоформами (см. Дополнительный файл 1; рисунок S2B, C).
Затем мы сравнили использование данных Iso-seq и RNA-seq для оценки экспрессии изоформ, сосредоточив внимание на подмножестве изоформ, идентифицированных с использованием обоих методов ( n = 19094). Мы рассчитали уровни экспрессии изоформ (tpm), используя
- 1)
Только данные Iso-seq (FL-TPM; полноразмерное чтение стенограммы на миллион)
- 2)
Данные РНК-seq с использованием транскриптомов, определенных Iso-seq (TPM; kallisto)
- 3)
Данные последовательности РНК с использованием транскриптомов с коротким чтением (TPM; kallisto)
Этот анализ показал, что уровень экспрессии, полученный из Iso-seq, лишь слабо коррелирует с уровнями экспрессии, полученными из РНК-seq (Pearson r = 0.27, Spearman r = 0,5), что, вероятно, связано с относительно меньшей глубиной секвенирования в Iso-seq. Однако уровни экспрессии транскриптов с использованием считываний РНК-seq, картированных на Iso-seq или транскриптомы короткого чтения, сильно коррелировали (Pearson r = 0,92, Spearman r = 0,87) (см. Дополнительный файл 1; рисунок S2D). Взятые вместе, эти анализы демонстрируют, что данные Iso-seq и RNA-seq дополняют друг друга — использование Iso-seq позволяет идентифицировать полноразмерные изоформы клеточной линии и транскриптомы GC, сводя к минимуму ошибки сборки и артефакты из-за неполных фрагментов, в то время как Использование коротко читаемых РНК-seq с высоким охватом позволяет более точно оценивать уровни экспрессии генов.
Чтобы дополнительно охарактеризовать долго читаемые транскрипты, мы сравнили как известные, так и новые изоформы различных типов событий сплайсинга. Используя инструмент SUPPA2 [22], мы количественно определили уровни удержания интрона (RI), пропуска экзона (SE), альтернативного 3′-акцептора (A3), альтернативного 5′-донора (A5), альтернативного первого экзона (AF), альтернативного события сплайсинга последнего экзона (AL) и взаимоисключающего экзона (MX) (рис. 3a). Всего было идентифицировано 63786 альтернативных событий сплайсинга (рис. 3b), в которых события AF внесли наибольший вклад (22399, 35.1%). События AF с большей вероятностью обнаруживались в новых изоформах по сравнению с известными изоформами (тест Фишера, значение p <2,2 × 10– 16 , отношение шансов = 2,4), что позволяет предположить, что использование альтернативных первых экзонов могло быть значительным. недооценена в предыдущих исследованиях с использованием данных коротко читаемых РНК-последовательностей. Эти данные также подтверждают предыдущие сообщения о том, что использование альтернативных промоторов является основным источником транскриптомного и функционального разнообразия при раке [8].
Рис. 3Виды альтернативных переделок сварки. a Классификация событий альтернативного сплайсинга (AS) с использованием SUPPA. A3 — Альтернативный 3′-сайт сращивания; A5 — Альтернативный 5′-сайт сплайсинга; AF — альтернативный первый экзон; AL — альтернативный последний экзон; MX — взаимоисключающий экзон; RI — удерживаемый интрон; SE — пропуск экзона. b Альтернативные промоторные сайты (AF) являются наиболее распространенным типом обнаруживаемых событий сплайсинга, и большинство событий AF обнаруживаются в новых изоформах (NIC или NNC). c Альтернативные события сплайсинга в новых изоформах экспрессируются на более низком уровне.Новые AF, AL и MX показывают относительно более высокую экспрессию. d Изменчивость процента сплайсинга событий SE (PSI), наблюдаемая по типам SE. Наибольшая вариабельность наблюдается в AF новых изоформ. e Анализ генной онтологии 500 наиболее вариабельных событий SE — гены, участвующие в обычно нерегулируемых путях при раке желудка, также являются мишенями альтернативного сплайсинга / промотора (например, клеточной адгезии и процессов развития). Значение p было скорректировано с учетом смещения длины гена. f Тепловая карта, показывающая 50 наиболее изменчивых событий AS — большинство наиболее изменчивых AS — это AF
Мы также использовали SUPPA2 для расчета относительных уровней экспрессии (представленных как «процент сращивания»; PSI) и вариации в выражении для различных сращиваний. классы. По линиям GC новые изоформы каждого класса сплайсинга демонстрировали более низкие уровни экспрессии по сравнению с известными изоформами (средний PSI 0,38 против 0,69 в известных изоформах; парные t -тест p , значение 6,4 × 10 — 4 ).Однако среди классов сплайсинга новые события AF, AL и MX (средний PSI 0,46–0,47) показали относительно более высокую экспрессию, чем другие типы событий сплайсинга (средний PSI 0,31–0,37; t -test p значение 1,4 × 10 — 3 ) (рис. 3в). Новые изоформы, как правило, выражались более вариабельно (среднее стандартное отклонение 0,15 против 0,12; парное t -тест p значение 1,5 × 10 — 3 ), причем события AF, AL и MX демонстрируют наибольшую дисперсию по линиям ( среднее стандартное отклонение 0.20 против 0,11; t -тест p значение 0,01) (рис. 3г). Иерархическая кластеризация по линиям показала, что наиболее вариабельные изоформы часто являются новыми изоформами, связанными с событиями ФП. Эти результаты были надежными, даже когда были проанализированы 500–1000 наиболее изменчивых событий сращивания (данные не показаны). Анализ генной онтологии изоформ, демонстрирующих первые 1000 событий сплайсинга с наибольшей дисперсией, показал, что эти изоформы обогащены путями, которые, как известно, не регулируются в GC, такими как процессы развития и клеточная адгезия (значения p с поправкой на длину гена 4.9 × 10 — 8 и 8,6 × 10 — 6 ) (рис. 3e), включая несколько известных генов рака, таких как MADD , PTK2 и NUMA1 (рис. 3f).
Длинночитываемые транскриптомы используются для анализа первичных профилей GC RNA-seq
Учитывая обогащение альтернативных промоторов новыми изоформами и их высокую вариабельность между образцами, мы подробно остановились на этом конкретном подклассе сплайсинга. Здесь мы применили proActiv [8], пакет R, который оценивает активность промотора на основе данных выровненных последовательностей РНК, примененных к эталонному транскриптому.Вкратце, proActiv количественно определяет экспрессию промотора с использованием набора уникальных считываний соединений, и мы ранее показали, что активность промотора, предсказанная proActiv , демонстрирует более высокую согласованность с данными гистонов CAGE и h4K4me3 при сравнении с другими методами. Чтобы оценить точность предсказаний промотора proActiv , мы коррелировали предсказанные активности промотора из стандартной РНК-seq с предсказанными транскриптомами, полученными из Iso-seq в различных диапазонах длин изоформ (дополнительный файл 2, таблица S3).Мы наблюдали более сильную корреляцию для более коротких изоформ по сравнению с более длинными изоформами. Однако важно отметить, что для всех категорий длины изоформ мы наблюдали значительно более высокую корреляцию между активностями промотора, полученными с помощью программного обеспечения Iso-seq и proActiv в одной и той же клеточной линии, по сравнению с активностями промотора, полученными на основе различных клеточных линий, что позволяет предположить, что активность промотора определяется с использованием . proActiv наиболее соответствует данным Iso-seq той же клеточной линии. Например, для изоформ с длиной гена менее 2 т.п.н. средний коэффициент корреляции между одними и теми же линиями был равен 0.63, по сравнению с 0,49 при сравнении между различными линиями клеток (рис. 4a; t -test p значение 1,8 × 10 — 12 ). Аналогичные корреляции наблюдались при ограничении этого анализа только известными или новыми изоформами (дополнительный файл 2, таблица S4). Умеренная корреляция, наблюдаемая между Iso-seq и proActiv из одних и тех же клеточных линий, вероятно, связана с относительно более низкой глубиной секвенирования и ошибками длины генов в методах Iso-seq. Подход с использованием полноразмерных изоформ Iso-seq для создания эталонного транскриптома и последующего количественного определения экспрессии изоформы с помощью короткого чтения также использовался другими специалистами в этой области [23,24,25,26].
Рис. 4Пейзаж использования альтернативных промоторов в данных Iso-seq. a Корреляционная матрица между детектированными изоформами из данных Iso-seq с промоторной активностью, оцененной из коротко читаемой РНК-seq Illumina для изоформ длиной менее 2 т.п.н. Наивысшие коэффициенты корреляции всегда наблюдаются между предсказанной активностью промотора по данным Iso-seq и Illumina для одной и той же клеточной линии. b Количество идентифицированных генов и промоторов Iso-seq.Двадцать пять процентов всех генов имеют несколько промоторов. c Пример известных и новых промоторов. Промоторы считаются известными, если по крайней мере один транскрипт FSM инициируется с сайта промотора, и новыми, если транскрипт FSM не инициируется с сайта промотора. Промотор гена с более высокой средней активностью дополнительно назначается как главный промотор, а все другие промоторы для того же гена назначаются как минорные промоторы. d Отнесение основного и второстепенного промотора к данным короткого считывания РНК-seq.Новые промоутеры часто бывают второстепенными. e Схематическое представление взаимосвязи между областями 5 ‘UTR, CD и 3’ UTR. В среднем использование альтернативных промоторов изменяет около 22–24% кодирующих областей. f Пример того, как использование альтернативного промотора может модифицировать CD ( PSMB4 ) и 5′-UTR ( MRPL28 )
По линиям GC мы идентифицировали 18 293 активных промотора, отображаемых на 13 143 гена (рис. 4b). Двадцать пять процентов генов (3257) были связаны как минимум с 2 отдельными промоторами.Мы классифицировали активные промоторы как основные и второстепенные в зависимости от их средней промоторной активности в 10 линиях клеток, а также классифицировали их как известные или новые промоторы (рис. 4c). Мы обнаружили, что промоторы, связанные с новыми изоформами, часто являются минорными промоторами (тест Фишера, p , значение <2,2 × 10 –16 ), которые экспрессируются на более низких уровнях (рис. 4d). Однако было предсказано, что 21% новых изоформ являются основным промотором в линиях GC (например, MIB2 , см.рис.4в). Чтобы понять последствия использования альтернативных промоторов в нижележащих функциональных областях, мы рассчитали общую долю 5′-нетранслируемых областей (UTR), областей кодирующей последовательности (CD) и 3’UTR между изоформами, инициированными разными промоторами. Мы обнаружили, что изменения в областях 5’UTR часто сопровождаются изменениями в нижележащих CD и 3’UTR, при этом большинство известных / новых пар промоторов демонстрируют потенциальные изменения в их составе CD (1734/2059; 84,2%). Средняя степень изменений, наблюдаемых для каждой пары промоторов (главный / минорный или известные / новые промоторы), составляла 22–24% для CD и 35% для 3’UTR (рис.4д). На рисунке 4f показаны примеры использования альтернативного промотора, связанного с последующим использованием отдельных CD и 3′-UTR-областей.
Чтобы подтвердить экспрессию новых изоформ на уровне белка, мы запросили собственный набор данных протеомики масс-спектрометрии из 10 линий клеток, проанализированных с помощью Iso-seq. Вкратце, предсказанные GeneMarkS-T последовательности, кодирующие белок для всех изоформ, были добавлены в базу данных последовательностей, кодирующих белок Gencode v32, для формирования эталонного протеома. Уникальные пептиды были идентифицированы с помощью MaxQuant [27] с использованием этого эталонного протеома.Этот протеомный анализ идентифицировал 930 уникальных пептидов из 428 белков Iso-seq, которых нет в базе данных Gencode v32 (дополнительный файл 2; таблица S5). Важно отметить, что мы смогли проверить несколько уникальных пептидных последовательностей, связанных с новыми промоторными сайтами (дополнительный файл 1; рисунок S3), что подтверждает идею о том, что многие новые изоформы Iso-seq действительно экспрессируются на пептидном уровне. Кроме того, мы также выполнили 5 ‘быструю амплификацию концов кДНК (RACE) для валидации двух новых изоформ ( FGFR4 , TMEM59 ; дополнительный файл 1; рисунок S4a) и валидацию вестерн-блоттингом новых изоформ ARID1A и TMEM59 (дополнительные Файл 1; рисунок S4b).Примечательно, что ранее сообщалось о 5′-RACE и экспрессии белка новой изоформы MET [28,29,30], что дополнительно подтверждает способность нашего конвейера идентифицировать новые ассоциированные с раком промоторы. Эти результаты предполагают, что использование альтернативного промотора может способствовать функциональной диверсификации протеома, позволяя одному гену выбирать несколько последовательностей, кодирующих белок.
Используя proActiv , мы затем расширили наши предсказания промотора на основе полноразмерных транскриптов до набора данных TCGA GC RNA-seq (282 рака желудка и 33 нормальных образца).Мы заметили, что активность промотора различается между опухолевыми и нормальными образцами, а также между различными молекулярными подтипами GC (рис. 5a). Затем мы применили DESeq2 для проведения анализа использования дифференциального промотора на опухолевых и нормальных образцах. Сравнение опухолевых и нормальных образцов выявило 2389 изоформ с повышенной и 2049 подавляемых изоформ в GC (FDR <1 × 10 -3 ; дополнительный файл 2, таблицы S6 и S7). Примечательно, что промоторы, активированные в GC ( n = 2389), значительно чаще имели изменения в своих CD (средняя степень измененных CD на пару промоторов, 27.5% против 20,9%; ( скорректированная по длине гена p значение 3,2 × 10 — 43 ) (рис. 5б). Мы наблюдали усиленные изоформы, содержащие новые изоформы промоторов известных онкогенов, такие как MET , FGFR4 и ERBB3 (рис.5в, справа). Повторение этого анализа во второй независимой когорте из 20 пар образцов рака желудка повторно идентифицировало MET и FGFR4 как активированные в образцах GC (рис. 5c, слева). Подавленные промоторы не были связаны с изменениями CD (21,8% против 20,9%; t -test p значение 0,42) (фиг. 5d).
Рис. 5Количественная оценка транскриптома Iso-seq в наборе данных рака желудка TCGA. — T-sne график активности промотора в 315 образцах TCGA рака желудка (282 опухоли и 33 нормальных). b Анализ генной онтологии активированных промоторов в наборе данных TCGA GC. Значение p было скорректировано с учетом смещения длины гена. c Вулкановый график, показывающий логарифмическое изменение активности промотора в образцах опухолей. Новые промоторы нескольких онкогенов рака желудка ( FGFR4 , MET и ERBB3 ) активированы в наборе данных TCGA (справа). В независимом наборе данных (20 пар T-N) также обнаружено, что новые промоторы MET и FGFR4 имеют повышенную регуляцию, в то время как новый промотор для ERBB3 существенно не повышается. d Измененные CD по статусу промоторной активности. Активированные промоторы имеют более крупное изменение CD. и Верхняя панель показывает изоформы гена, инициированные из известных и новых промоторов FGFR4 , MET и ERBB3 . Нижняя панель показывает активность промотора и частоту обнаружения в нормальных образцах и подтипах TCGA рака желудка. Новые промоторы экспрессируются на более низком уровне, но имеют более высокую повышающую регуляцию и экспрессируются в большем количестве образцов опухоли по сравнению с нормальными образцами. f Верхняя панель показывает белковые домены изоформ, инициированных из известных и новых промоторов FGFR4 , MET и ERBB3 . Нижняя панель показывает предсказание сигнального пептида в транскриптах, инициированных с известных и новых промоторных сайтов, демонстрируя потерю последовательностей сигнального пептида в новых изоформах
Три новых изоформы MET , FGFR4 и ERBB3 предположительно инициируются от промоторов, отличных от аннотированных TSS (на расстоянии 51 т.п.н. ( MET ), 438 п.н. ( FGFR4 ), 248 п.н. ( ERBB3 ), и в большей степени активированы в образцах опухолей по сравнению с их известными изоформами (рис.5д). Как и в общей популяции, промоторная активность и частота обнаружения (суррогатная степень экспрессии) были ниже для этих новых изоформ по сравнению с известными изоформами, а новые изоформы демонстрировали большую вариабельность между опухолями. Например, новая изоформа MET высоко экспрессируется в подтипе CIN (log2-кратное изменение = 2,1, p значение = 5,5 × 10 — 16 ), но обеднена EBV (log2-кратное изменение = -4,7, ). p = 9,4 × 10 -15 ) по сравнению с другими образцами ГХ.Сравнение CD показало, что наиболее функционально важные белковые домены сохраняются в новых изоформах; однако все три новые изоформы показали усечение N-концевого белка и удаление домена Sema в случае MET (фиг. 5f) . Предполагается, что все три новые изоформы, инициированные из новых промоторных сайтов, нарушают последовательности сигнального пептида, необходимые для локализации на клеточной мембране. Сообщалось о сходных механизмах, при которых использование альтернативных промоторов приводит к локализации белка в другом клеточном компартменте [31].Взятые вместе, эти наблюдения предполагают, что новые промоторные сайты могут позволить генам приобретать новые функциональные роли и регулироваться специфическим для подтипа образом.
Затем мы запросили ReMap [32], чтобы идентифицировать TF, обогащенные опухолеспецифическими промоторами. Атлас ReMap 2018 содержит пики регуляторов транскрипции, полученные из курируемых баз данных ChIP-seq, ChIP-exo DAP-seq и ENCODE, извлеченных из GEO (Gene Expression Omnibus), содержащих 485 регуляторов транскрипции для 346 типов клеток человека из 2829 наборов данных ChIP-seq.Мы интегрировали профили занятости 485 TF против альтернативных промоторов с повышенной регуляцией (FDR <0,001; n = 2389) по сравнению со всеми промоторами, идентифицированными в этом исследовании ( n = 18 293), используя пакет ReMapEnrich R (https: // github. com / remap-cisreg / ReMapEnrich). Из 485 ТФ, доступных в Remap, было обнаружено, что 204 ТФ значительно увеличены ( q <0,001) на активированных промоторах, по крайней мере, в одном эксперименте с ChIP-seq. Чтобы оценить надежность нашего анализа, мы также выполнили тот же анализ, используя другую базу данных прямого взаимодействия TF-ДНК (UniBind).UniBind содержит информацию о 231 TF из наборов данных ChIP-seq 1983 года. Четыре из 10 TF с наивысшим рейтингом от ReMap были также предсказаны инструментом UniBind_enrichment (https://unibind.uio.no/enrichment/), включая E2F4, E2F1, MYC и MXI1 (дополнительный файл 1; рисунок S5a, S5b. ). Это может указывать на возможные TF, регулирующие использование альтернативных промоторов в GC.
Мы также исследовали промоторы на предмет изменений в метилировании ДНК, используя MeDIP-seq по всему геному (зависимая от метилирования иммунопреципитация с последующим секвенированием) для 9/10 клеточных линий.Вкратце, считывания MeDIP выравнивали с геномом человека с помощью bwa, а дубликаты удаляли с помощью samtools. Пики метилирования ДНК выявляли с помощью MACS2 с контролем ввода. В клеточных линиях 7/9 мы наблюдали, что неэкспрессирующие изоформы (измеренные с использованием данных Iso-seq), как правило, имеют более высокие уровни метилирования вблизи их промоторной области. Напротив, экспрессируемые изоформы имели тенденцию к снижению уровней метилирования ДНК — эта корреляция наблюдалась как для известных, так и для новых промоторов (дополнительный файл 1; рис. S6), что дает дополнительные доказательства того, что новые экспрессируемые промоторы являются промоторами bona-fide , поскольку они проявляют сходные эпигенетические признаки с известными экспрессируемыми промоторами.
Клинический результат, связанный с использованием альтернативных промоторов
Поскольку новые промоторы экспрессируются по-разному, мы исследовали, может ли их использование выявить новые биомаркеры клинического исхода в GC. Чтобы проверить эту возможность, мы сопоставили различные паттерны активности промотора с выживаемостью без прогрессирования в наборе данных TCGA (рис. 6a). Образцы опухолей были стратифицированы на использование высокого и низкого промотора на основе оптимальных пороговых значений, определенных с использованием функции выживаемости-точки отсечения, и продолжительности выживания в двух группах, сравниваемых с использованием теста логарифмического ранга.
Рис. 6Клинический результат, связанный с использованием альтернативных промоторов. a Диаграмма рассеяния скорректированных значений p для генов с известными и новыми промоторами. Примеры прогностического гена (синий), прогностического известного промотора (зеленый) и нового прогностического промотора (красный) включают TFPI , KRT7 и KDM4A соответственно. b График Beeswarm, показывающий среднюю корреляцию между известными и новыми промоторами.Прогностических промоторов (известных или новых) больше, чем прогностических генов, а прогностические промоторы имеют более низкую корреляцию, чем прогностические гены, что позволяет предположить, что отдельные промоторы регулируются независимо. c Графики выживаемости, показывающие значительную связь новых и известных промоторов KDM4A , TFPI и KRT7 с выживаемостью без прогрессирования. d Процент известных и новых промоторов, которые по-разному экспрессируются в прогностических генах, прогностических промоторах и непрогностических генах.Дерегулированная экспрессия генов и изоформ является прогностическим фактором выживания. и Коробчатые диаграммы общего и частного процента CD в прогностических генах, прогностических промоторах и непрогностических генах. Прогностические промоторы обогащены промотор-специфическими областями CD. f Генная структура известного и нового промоторов ARID1A на уровне транскрипта и белка. Кривая выживаемости показывает связь между известными и новыми промоторами ARID1A с выживаемостью без прогрессирования при раке желудка
В 1783 генах с предсказанными известными и новыми промоторами мы идентифицировали 871 ген с прогностическими промоторами (FDR <0.05). Из них 202 гена (23%) были прогностическими как для известных, так и для новых промоторов, и, как и ожидалось, для этих генов промоторная активность известных и новых промоторов сильно коррелировала (средняя корреляция 0,43). Напротив, гены 435 и 234 были прогностическими только для известного или нового промотора соответственно, и для этих генов промоторная активность известных и новых промоторов была несвязанной (средняя корреляция 0,28, t -test, p значение 1,6 × 10 — 11 ) (рис.6б). Примеры генов с известными прогностическими промоторами (например, KRT7 ), прогностических генов (например, TFPI ) и новых прогностических промоторов (например, KDM4A ) показаны на фиг. 6c. По сравнению с непрогностическими генами, промоторы для прогностических генов и сами промоторы с большей вероятностью будут дифференциально экспрессироваться в GC (тест Фишера, p , значение 1,5 × 10 -13 , отношение шансов = 1,8) (рис. 6d). Интересно, что мы обнаружили, что области CD, специфичные для изоформ, связанные с новыми промотор-специфическими областями CD, специфически обогащены генами с новыми прогностическими промоторами (тест Краскела-Уоллиса, p , значение 2.1 × 10 -7 ) (рис. 6e), предполагая, что усиление CD из-за использования новых промоторных сайтов может наделять гены дополнительными ролями. Напротив, была значительно более слабая ассоциация для изоформ-специфических областей CD, связанных с известными промоторами (тест Краскела-Уоллиса, p , значение 0,05). Этот анализ также подтверждает гипотезу о том, что новые промоторы могут регулироваться независимо и что они могут иметь различные роли от известных аналогов.
В качестве примера мы обнаружили новую изоформу ARID1A , которая значительно снижает выживаемость без прогрессирования заболевания (рис.6е). Мы идентифицировали 3 изоформы транскрипта ARID1A и 2 промотора из каталога полноразмерных изоформ GC. Обнаруженные новые и известные изоформы белка ARID1A , как предполагается, усекают первые 384 и 274 N-концевые аминокислоты соответственно из канонического белка (NP_006006.3). Изоформы белка, соответствующие каноническому транскрипту ARID1A в Ensembl (транскрипт Ensembl с самой длинной трансляцией CD), не были обнаружены в нашем наборе данных.Интересно, что высокая экспрессия нового промотора связана с более низкой выживаемостью (логарифмический ранг p , значение 2,1 × 10 -7 ), тогда как известный промотор не имеет значимой связи (логарифмический ранг p , значение 0,09). Известный промотор ARID1A значительно истощен в подтипе MSI (log2-кратное изменение = — 0,35, p значение = 3,2 × 10 — 3 ). Напротив, новый ARID1A не выражается по-разному в MSI (log2-кратное изменение = 0.04, p значение = 0,88), но показывает пограничное подавление (log2-кратное изменение = — 0,42, p значение 4,2 × 10 — 2 ) в подтипе CIN. Дальнейшие исследования необходимы для выяснения функциональной роли различных изоформ ARID1A .
Сравнение длинных методов секвенирования и сборки генома растений | GigaScience
Аннотация
Предпосылки
Технологии секвенирования продвинулись до такой степени, что стало возможным генерировать высокоточные сборки хромосомного масштаба с разрешенными гаплотипами.Доступно несколько технологий секвенирования с длинным считыванием, и было разработано все больше алгоритмов для сборки считываний, сгенерированных этими технологиями. Поэтому при запуске нового проекта генома сложно выбрать наиболее экономичную технологию секвенирования, а также наиболее подходящее программное обеспечение для сборки и полировки. Таким образом, важно сравнить различные подходы, применяемые к одной и той же выборке.
Результаты
Здесь мы представляем сравнение 3 технологий долгого чтения, примененных к сборке de novo генома растения, Macadamia jansenii .Мы создали данные секвенирования с использованием технологий Pacific Biosciences (Sequel I), Oxford Nanopore Technologies (PromethION) и BGI (чтение длинных фрагментов в одной пробирке) для одного и того же образца. Несколько ассемблеров были протестированы при сборке журналов Pacific Biosciences и Nanopore reads. Также представлены результаты, полученные в результате объединения технологий длительного чтения или технологий короткого и длительного чтения. Сборки сравнивались на предмет смежности, базовой точности и полноты, а также затрат на секвенирование и требований к материалам ДНК.
Выводы
Три технологии длительного чтения привели к получению очень смежных и полных геномных сборок M. jansenii . Во время секвенирования стоимость каждого метода значительно отличалась, но постоянное совершенствование технологий привело к повышению точности, увеличению пропускной способности и снижению затрат. Мы предлагаем регулярно обновлять это сравнение отчетами о значительных итерациях технологий секвенирования.
Введение
Достижения в области секвенирования ДНК позволяют проводить быстрый анализ геномов, способствуя биологическим открытиям.Секвенирование сложных геномов, которые очень велики и имеют большое количество повторяющихся последовательностей или многих копий подобных последовательностей, остается сложной задачей. Геномы многих растений сложны, и качество опубликованных последовательностей остается относительно низким. Однако улучшения в секвенировании с длинным считыванием упрощают создание высококачественных последовательностей для сложных геномов.
Теперь мы представляем сравнение 3 методов секвенирования с длительным считыванием, примененных к секвенированию de novo растения, Macadamia jansenii .Это редкий вид, который является близким родственником ореха макадамия, недавно одомашненного на Гавайях и в Австралии. В дикой природе он растет как многоствольное вечнозеленое дерево, достигающее 6–9 м в высоту с листьями, имеющими полные края и обычно в 3 мутовках. Орехи небольшие (диаметр 11–16 мм) и гладкие, твердые, коричневая оболочка, покрывающая кремовое глобулозное ядро, горькое и несъедобное [1]. Этот вид был обнаружен как единая популяция из ~ 60 растений в дикой природе в восточной Австралии [2].Это цветковое растение (покрытосеменное) семейства Proteaceae, которое является базальным по отношению к большой ветви эвдикота филогении цветковых растений [3]. Геномы этой группы плохо охарактеризованы, при этом наиболее хорошо секвенированные геномы растений представляют собой центральные эвдикоты или однодольные растения, имеющие экономическое значение [4]. Знание генома этого вида поддержит усилия по сохранению исчезающих видов в дикой природе и выявлению новых черт, таких как небольшой рост, для использования в селекции растений.Секвенирование диких родственников сельскохозяйственных культур является неотложной задачей, поскольку многие популяции имеют решающее значение для диверсификации генетики сельскохозяйственных культур для обеспечения продовольственной безопасности в ответ на изменение климата [5], но им также угрожает исчезновение из-за изменений в землепользовании или климате [6].
Род macadamia включает 4 вида: Macadamia integrifolia, Macadamia tetraphylla, Macadamia ternifolia и Macadamia jansenii . Сорта макадамии являются диплоидными (2n = 28), с оценками размера генома k на основе -меров в диапазоне от 758 МБ для M.tetraphylla [7] до 896 МБ для M. integrifolia [8]. Первый черновой вариант сборки генома широко выращиваемого сорта M. integrifolia HAES 741 был сконструирован из данных коротко читаемых последовательностей Illumina и был сильно фрагментирован (518 МБ, 193 493 каркаса, N50 = 4 745 п.н.) [9]. Улучшенная сборка HAES 741 была создана с использованием комбинации длинночитываемых данных Pacific Biosciences (PacBio) и данных последовательностей Illumina с парным концом (745 МБ, 4094 каркаса, N50 = 413 т.п.н.) [8]. Сборка генома M.tetraphylla также недавно получили с использованием комбинации длинных данных Oxford Nanopore Technologies (ONT) и коротких данных последовательностей Illumina (751 МБ, 4335 контигов, N50 = 1,18 МБ) [7].
Секвенирование с длинным чтением дает данные, которые облегчают сборку генома, чем это возможно с коротким чтением [10–12]. Длина и качество последовательности, обеспечиваемые доступными платформами секвенирования, продолжали улучшаться. Произведенные чтения могут быть использованы для сборки контигов или в качестве каркаса для сборки контигов, созданных с помощью этих методов или из коротких чтений [13].В настоящее время PacBio и ONT являются наиболее часто используемыми технологиями для генерации длинных операций чтения. Одномолекулярное секвенирование в реальном времени (SMRT), разработанное PacBio, может генерировать считывания в десятки килобаз с использованием режима непрерывного долгого считывания, что обеспечивает высококачественную сборку генома de novo . ONT обеспечивает прямое секвенирование в реальном времени длинных фрагментов ДНК или РНК, анализируя нарушение электрического тока, вызванное молекулами при их движении через нанопоры белка. Совсем недавно BGI представила технологию чтения длинных фрагментов с одной пробиркой (stLFR) [14] в качестве альтернативы генерации настоящих длинных считываний.stLFR основан на совместном штрих-кодировании ДНК [15,16], то есть добавлении одной и той же последовательности штрих-кода к субфрагментам исходной длинной молекулы ДНК. В процессе stLFR поверхности микрошариков используются для создания миллионов миниатюрных реакций штрих-кодирования в одной пробирке. Важно отметить, что stLFR обеспечивает совместное штрих-кодирование почти одной молекулы за счет использования большого количества микрогранул и комбинаторного процесса для создания ~ 3,6 миллиарда уникальных последовательностей штрих-кода. По этой причине ожидается, что это позволит получить высококачественные и почти завершенные сборки de novo .Здесь мы сравниваем данные Sequel I (PacBio), PromethION (ONT) и stLFR (BGI) для одного и того же образца ДНК и оцениваем качество сборок, которые могут быть созданы непосредственно из этих наборов данных.
Методы
Растительный материал
Молодые листья (40 г) M. jansenii были получены с дерева под номером 1005 и размещены в Исследовательском центре Маручи, Министерство сельского хозяйства и рыболовства, Намбор 4560, Квинсленд, Австралия.Образец M. jansenii , использованный в этих экспериментах, представлял собой клонально размноженное дерево ex situ , посаженное в дендрарии исследовательского центра Maroochy. Ни один из листьев, используемых в этих экспериментах, не был собран с диких деревьев in situ. Молодые листья собирали, помещали на лед в мешки и в течение 3 часов быстро замораживали в жидком азоте и хранили при -20 ° C до дальнейшей обработки для измельчения тканей с использованием ступки и пестика или смесительной мельницы, как описано ниже.
Экстракция геномной ДНК
Ткань листа (10 г) сначала грубо измельчали в жидком азоте с помощью ступки и пестика. Затем ступку и пестик с грубо измельченной тканью с остаточным жидким азотом помещали на сухой лед. Этот этап обеспечивал поддержание температуры грубо измельченной ткани около -80 ° C, позволяя жидкому азоту полностью испариться, что является важным требованием на этапе измельчения. Грубо измельченную ткань листа измельчали до мелкого порошка в стальных емкостях объемом 50 мл, используя Mixer Mill MM400 (Retsch, Германия).Измельченную в порошок ткань листа хранили при -20 ° C до тех пор, пока не потребуется дальнейшая экстракция ДНК. Геномную ДНК (гДНК) выделяли из измельченной ткани листа согласно [17] с некоторыми модификациями. Используя охлаждаемый жидким азотом шпатель, замороженную измельченную ткань листа (3 г) добавляли в пробирки объемом 50 мл (Corning или Falcon), содержащие теплый (40 ° C) буфер для ядерного лизиса (8 мл) и 5% раствор саркозила (5 мл). ). Пробирки инкубировали при 40 ° C в течение 45 мин с периодическим (каждые 5 мин) осторожным перемешиванием путем переворачивания пробирок.РНК расщепляли добавлением раствора РНКазы (10 мг / мл), содержимое осторожно перемешивали, переворачивая пробирки, с последующей инкубацией при комнатной температуре в течение 10 мин. Две экстракции хлороформом проводили следующим образом. В пробирки добавляли хлороформ (10 мл) и осторожно перемешивали, переворачивая пробирки 50 раз. Пробирки центрифугировали при 3500 g в течение 5 минут в роторе с откидным ковшом. Супернатант переносили в свежие пробирки объемом 50 мл и дважды повторяли экстракцию хлороформом. Супернатант переносили в свежие пробирки на 50 мл и ДНК осаждали изопропанолом.На каждый 1 мл супернатанта добавляли 0,6 мл изопропанола и осторожно перемешивали содержимое, переворачивая пробирки 20–25 раз. Пробирки инкубировали при комнатной температуре в течение 15 минут, а затем центрифугировали при 3 500 g в течение 5 минут в роторе с откидным ковшом. Супернатант отбрасывали, а осадок ДНК промывали от любых соосажденных солей, добавляя 10 мл 70% этанола и инкубируя пробирки при комнатной температуре в течение 30 мин. Пробирки центрифугировали при 3500 g в течение 5 минут в роторе с выдвижным ведром, супернатант отбрасывали, а осадок ДНК полусушили для удаления любых остатков 70% этанола, инкубируя пробирки в течение 10 минут в перевернутом виде над фильтровальной бумагой. .ДНК растворяли, добавляя 100 мкл буфера ТЕ, а затем при необходимости добавляя дополнительные 50 мкл буфера ТЕ. Раствор ДНК переносили в пробирки объемом 2 мл, свободные от нуклеаз, а затем центрифугировали при 14000 g в течение 45 мин в настольной центрифуге. Супернатант осторожно переносили в свежие пробирки на 2 мл, качество проверяли на спектрофотометре, и ДНК разделяли на 0,7% -ном агарозном геле. Затем ДНК хранили при -20 ° C до использования для секвенирования.
Подготовка и секвенирование библиотеки гДНК PacBio
Библиотеки секвенирования ДНКбыли подготовлены с использованием Template Prep Kit 1.0-SPv3 (PacBio, 100-991-900) в соответствии с протоколом для библиотек SMRTbell> 30 kb (PacBio, Part No. PN 101-024-600 Version 05). Геномную ДНК (15 мкг) не фрагментировали, а просто очищали с помощью гранул AMPure PB. Очищенную гДНК (10 мкг) обрабатывали экзонуклеазой VII с последующей реакцией восстановления повреждений ДНК, реакцией восстановления конца и очисткой гранулами AMPure PB. Адаптеры были лигированы к очищенным фрагментам ДНК с тупыми концами при инкубации в течение ночи. Образец с лигированным адаптером обрабатывали экзонуклеазой III и экзонуклеазой VII для удаления неудавшихся продуктов лигирования с последующей очисткой гранулами AMPure PB.Размер очищенного образца был выбран с использованием Blue Pippin с кассетой 0,75% агарозы без красителей и маркером U1 (Sage Science, BUF7510, Mulgrave, Виктория, Австралия) и 0,75% DF Marker U1 high-pass 30-40 kb vs3 запустить протокол с отсечкой BPstart 35000 п.н. После выбора размера образцы очищали гранулами AMPure PB с последующей реакцией восстановления повреждений ДНК и окончательной очисткой гранулами AMPure PB. Конечная очищенная библиотека выбранного размера была количественно определена на флуорометре Qubit с использованием набора для анализа дцДНК HS Qubit (Invitrogen, Q32854, Thermo Fisher Scientific, Scoresby, Виктория, Австралия) для оценки концентрации и 0.4% агарозный гель Megabase (BioRad, 1613108, Gladesville, Новый Южный Уэльс, Австралия) для оценки размера фрагмента. Секвенирование выполняли с использованием PacBio Sequel I (PacBio Sequel System, RRID: SCR_017989) (программное обеспечение / химия v6.0.0). Библиотека была подготовлена для секвенирования в соответствии с калькулятором установки образца SMRT Link, следуя стандартному протоколу для диффузионной загрузки с очисткой гранул AMPure PB, с использованием Sequencing Primer v3, Sequel Binding Kit v3.0 и Sequel DNA Internal Control v3.Связанную с полимеразой библиотеку секвенировали на 8 ячейках SMRT с продолжительностью просмотра 10 часов с использованием набора Sequel Sequencing Kit 3.0 (PacBio, 101-597-900, Mulgrave, Виктория, Австралия) и Sequel SMRT Cell 1M v3 (PacBio, 101- 531-000, Малгрейв, Виктория, Австралия). Подготовка библиотеки и секвенирование были выполнены в Институте молекулярной биологии секвенирования (Университет Квинсленда).
Подготовка и секвенирование библиотеки ONT
Качество образца ДНК оценивалось в NanoDrop, Qubit и системе Agilent 4200 TapeStation.Образец ДНК секвенировали на ONT MinION (MinION, RRID: SCR_017985) и PromethION (PromethION, RRID: SCR_017987). Библиотеку MinION получали из 1500 нг входящей ДНК с использованием набора для секвенирования лигирования (SQK-LSK109, ONT, Оксфорд, Великобритания) в соответствии с протоколом производителя, за исключением того, что реакции восстановления и подготовки концов и период лигирования были увеличены до 30 мин. Реагенты сторонних производителей NEBNext end repair / dA-tailing Module (E7546), NEBNext, фиксированная формалином, парафиновая смесь для восстановления ДНК (M6630) и NEB Quick Ligation Module (E6056) были использованы во время подготовки библиотеки.Образец ДНК с лигированным адаптером количественно оценивали с использованием набора Qubit TM dsDNA HS Assay Kit (Thermo Fisher Scientific, Скорсби, Виктория, Австралия). Проточная кювета MinION R9.4.1 (FLO-MIN106, ONT, Оксфорд, Великобритания) была праймирована в соответствии с инструкциями производителя перед загрузкой смеси библиотек (75 мкл), содержащей 438 нг ДНК с лигированием адаптера, 25,5 мкл LB (SQK-LSK109 , ONT, Оксфорд, Великобритания) и 37,5 мкл SQB (SQK-LSK109, ONT, Оксфорд, Великобритания). Секвенирование MinION было выполнено с использованием MinKNOW (v1.15.4) и стандартного 48-часового сценария выполнения.Перед подготовкой библиотеки PromethION короткие фрагменты ДНК (<10 т.п.н.) сначала были истощены из образца ДНК (9 мкг), как описано в инструкциях производителя для набора Short Read Eliminator (SRE) (SKU SS-100-101-01, Circulomics Inc, Балтимор, Мэриленд, США). Библиотеку PromethION получали из 1200 нг SRE-обработанной ДНК с использованием набора для секвенирования лигирования (SQK-LSK109, ONT, Oxford, UK). Все этапы приготовления библиотеки были такими же, как и при приготовлении библиотеки MinION, за исключением того, что ДНК с лигированной адаптером элюировали в 25 мкл буфера для элюции.Проточная кювета PromethION (FLO-PRO002, ONT, Оксфорд, Великобритания) была праймирована в соответствии с инструкциями производителя перед загрузкой библиотечной смеси (150 мкл), содержащей 390 нг ДНК с лигированной адаптером (24 мкл), 75 мкл SQB и 51 мкл LB (SQK-LSK109, ONT, Оксфорд, Великобритания). Секвенирование выполнялось с использованием MinKNOW (v3.1.23) и стандартного 64-часового сценария выполнения. Цикл секвенирования был остановлен через 21 час и была проведена промывка нуклеазой для восстановления забитых пор. Смесь для промывки нуклеаз готовили путем смешивания 380 мкл буфера для промывки нуклеаз (300 мМ KCl, 2 мМ CaCl 2 , 10 мМ MgCl 2 , 15 мМ HEPES pH 8) и 20 мкл ДНКазы I (M0303S, NEB, Ноттинг-Хилл, Виктория, Австралия).Смесь для промывки нуклеазами загружали в проточную кювету и инкубировали в течение 30 мин. Затем проточную кювету примировали, как указано выше, и загрузили свежей библиотечной смесью (150 мкл), содержащей 390 нг ДНК с лигированной адаптерами, и стандартный 64-часовой сценарий запуска был повторно запущен с использованием MinKNOW. Дозаправку цикла секвенирования выполняли каждые 24 часа путем добавления 150 мкл разбавленного SQB (1: 1, SQB: вода, свободная от нуклеаз) для поддержания стабильной скорости транслокации при секвенировании. Чтения ONT fast5 вызывались базой с использованием Guppy v3.0.3 с конфигурационным файлом dna_r9.4.1_450bps_hac_prom.cfg (PromethION) или dna_r9.4.1_450bps_hac.cfg (MinION) и параметрами —qscore_filtering -q 0 —recursive —device «cuda: 0 cuda: 1 cuda: 2 cuda: 3 «.
Подготовка и секвенирование библиотеки BGI stLFR
Библиотеки секвенирования stLFR получали с использованием набора для подготовки библиотек MGIEasy stLFR (MGI, Шэньчжэнь, Китай) в соответствии с протоколом производителя. Вкратце, образцы геномной ДНК были серийно разведены, а затем количественно определены с использованием набора для анализа дцДНК BR Qubit TM (Invitrogen, Карлсбад, Калифорния) и набора Qubit TM dsDNA HS Assay Kit (Invitrogen, Карлсбад, Калифорния) для более точной количественной оценки. результат.Для приготовления библиотеки использовали примерно 1,5 нг исходных молекул геномной ДНК. На первом этапе транспозоны, состоящие из последовательности захвата и последовательности распознавания транспозазы, вставляли через равные промежутки времени вдоль молекул гДНК. Затем эти молекулы ДНК со встроенным транспозоном гибридизовали с меченными штрих-кодом магнитными шариками диаметром 3 мкм, содержащими олигонуклеотидные последовательности, с сайтом отжига праймера ПЦР, штрих-кодом stLFR и последовательностью, комплементарной последовательности захвата на транспозоне.После гибридизации штрих-код переносили на субфрагменты ДНК, в которые вставлен транспозон, посредством стадии лигирования. Затем избыток олигонуклеотидов и транспозонов переваривали экзонуклеазой, а фермент транспозазы денатурировали додецилсульфатом натрия. Затем второй адаптер был введен с помощью ранее описанного лигирования 3′-ответвлений с использованием лигазы Т4 [18]. Наконец, была проведена ПЦР-амплификация с использованием праймеров, отжигаемых с последовательностями адаптеров 5′-гранул и 3′-ответвлений. Реакцию ПЦР очищали с использованием гранул Agencourt® AMPure XP (Beckman Coulter, Brea, CA) и количественно оценивали с использованием набора для анализа дцДНК HS Qubit TM (Invitrogen, Carlsbad, CA).Размеры фрагментов продукта ПЦР оценивали с помощью набора Agilent High Sensitivity DNA Kit (Agilent, 5067-4626) на биоанализаторе Agilent 2100. Средний размер фрагмента полученной библиотеки stLFR составлял 1003 п.н. Количество 20 нг продукта ПЦР из библиотеки stLFR использовали для приготовления ДНК-наношаров (DNB) с использованием набора для высокопроизводительного секвенирования stLFR DNBSEQ-G400RS (MGI, Шэньчжэнь, Китай) в соответствии с протоколом производителя. Подготовленную библиотеку DNB загружали на 2 дорожки проточной кюветы DNBSEQ-G400RS (MGI, Шэньчжэнь, Китай), а затем секвенировали на DNBSEQ-G400RS (MGI, Шэньчжэнь, Китай) с использованием набора для секвенирования stLFR DNBSEQ-G400RS (MGI, Шэньчжэнь). , Китай).Подготовка библиотеки и секвенирование были выполнены в Центре секвенирования BGI Australia (Центр исследования рака Клайва Бергхофера, Херстон, Квинсленд) и BGI-Shenzhen (Шэньчжэнь, Китай).
Секвенирование Illumina
Библиотека Illumina была приготовлена с использованием набора ДНК Nextera Flex. Библиотеку секвенировали на проточной кювете SP (14%) платформы секвенирования Illumina Nova Seq 6000 (Ramaciotti Center, Университет Нового Южного Уэльса, Австралия) с использованием протокола парных концов, чтобы произвести 112 миллионов считываний по 150 пар оснований. примерно 43-кратный охват генома.Средний размер вставки составил 713 п.н.
Подготовка к чтению последовательности
Длина и качество чтенияONT рассчитывались с помощью NanoPlot v1.22 [19]. Длинные чтения из PacBio и ONT были подготовлены с использованием 2 или 3 альтернативных стратегий соответственно:
Все: без фильтрации чтения
Отфильтровано: длинные чтения ONT были обрезаны адаптером с помощью Porechop v0.2.4 (Porechop, RRID: SCR_016967) [20]. Считывания ONT и PacBio фильтровались с помощью Filtlong v0.2.0 [21] путем удаления 10% наихудших чтений и чтений короче 1 КБ.
Пройден (только ONT): использовались только пройденные чтения (средняя оценка качества базового вызова> 7).
Субпотоки PacBio были случайным образом разбиты на подвыборку до 32-кратного покрытия генома с использованием Rasusa v0.1.0 [22]. Необработанные короткие чтения Illumina и BGI были обрезаны адаптером с помощью Trimmomatic v0.36 (Trimmomatic, RRID: SCR_011848) [23] (LEADING: 3 TRAILING: 3 SLIDINGWINDOW: 4: 15 ILLUMINACLIP: 2: 30: 10 MINLEN: 36).Обрезка хвоста PolyG была выполнена на считывателях Illumina с использованием fastp v0.20.0 (fastp, RRID: SCR_016962) [24].
Оценка размера генома
Подсчет k -меров с использованием обрезанных показаний Illumina и BGI был выполнен с использованием Jellyfish v2.210 (Jellyfish, RRID: SCR_005491) [25], генерируя частотные распределения k -меров 21, 23 и 25 -меры. Гистограммы вхождений k -меров были обработаны программой GenomeScope (GenomeScope, RRID: SCR_017014) [26], которая оценила размер гаплоида генома в 653 и 616 мегабайт с ~ 71% и 74% уникального содержания и уровня гетерозиготности. из 0.65% и 0,77% от показателей Illumina и BGI соответственно.
Сборка геномов
De novo сборка считываний ONT и PacBio была выполнена с использованием Redbean v2.5 (WTDBG, RRID: SCR_017225) [27], Flye v2.5 (Flye, RRID: SCR_017016) [28], Canu v1.8 (ONT ) или v1.9 (PacBio) (Canu, RRID: SCR_015880) [29] и Raven v1.1.6 [30] с параметрами по умолчанию. Для Redbean, Flye и Canu предполагаемый размер генома был установлен в 780 мегабайт [31]. Для данных ONT было выполнено 4 раунда исправления ошибок с помощью Racon v1.4.9 (Racon, RRID: SCR_017642) [32] с рекомендованными параметрами (-m 8 -x -6 -g -8 -w 500) на основе перекрытий minimap2 v2.17-r943-dirty [33], за которыми следует 1 раунд Medaka v0.8.1 [34] с использованием модели r941_prom_high для создания согласованной последовательности. Полученная последовательность была отполирована с помощью Pilon v1.23 (Pilon, RRID: SCR_014731) [35] с использованием считываний Illumina, сопоставленных с BWA-MEM v0.7.13 (BWA, RRID: SCR_010910) [36] и с настройками для исправления баз ( —фиксировать базы). Полировка согласованной последовательности Medaka с помощью считываний Illumina также была выполнена с помощью NextPolish v1.1.0 [37] с настройками по умолчанию (BWA для шага сопоставления). Гибридная сборка была создана с помощью MaSuRCA v3.3.3 (MaSuRCA, RRID: SCR_010691) [38] с использованием Illumina и ONT или PacBio для чтения и использования Flye v2.5 для выполнения окончательной сборки исправленных мегапространств (параметр FLYE_ASSEMBLY = 1). Сборка генома Diploid de novo считываний PacBio была выполнена с помощью FALCON v1.3.0 (FALCON, RRID: SCR_016089) [39] с использованием размера генома 780 МБ, отсечения длины 40 740 пар оснований и отсечения покрытия для чтения семян. -выкл 30.Всего было сгенерировано 19 Гб предварительно собранных чтений (24-кратное покрытие). После сборки и разделения гаплотипов с помощью FALCON-Unzip v1.2.0 [39] полировка была выполнена как часть рабочего процесса FALCON-Unzip. Чтения PacBio были сопоставлены с основной сборкой FALCON-Unzip с помощью minimap2 v2.17-r954-dirty [33]. Гистограмма охвата считыванием была сгенерирована из этого выравнивания с использованием Purge Haplotigs v.1.1.0 [40] для получения значений отсечки глубины считывания (-l 17 -m 52 -h 190), необходимых для идентификации избыточных контигов. Чтения Illumina были собраны с использованием SPAdes v3.13.1 (SPAdes, RRID: SCR_000131) [41].
Две дорожки чтения stLFR для одного и того же образца были демультиплексированы с использованием подфункции SuperPlus v1.0 [42] и объединены для последующего анализа. Последовательности адаптеров были удалены из считываемых данных с помощью Cutadapt v2.4 (cutadapt, RRID: SCR_011841) [43] с рекомендованными параметрами (—no-indels -O 10 —discard-trimmed -j 42). Затем последовательности считывания были преобразованы в формат 10X Genomics с помощью внутреннего программного обеспечения BGI, которое содержит 3 шага: (i) Изменение формата заголовка считывания с MGI на Illumina.(ii) Измените число качества основания «N» с 33 (код ASIC II =!) на 35 (код ASIC II = #), чтобы соответствовать системе качества 10X Genomics. (iii) Случайное объединение 2 или более штрих-кодов в 1 штрих-код из-за ограничения типов штрих-кодов для 10X Genomics. Чтобы удовлетворить требования ассемблера к памяти, штрих-коды с <10 чтениями были удалены из набора данных. Сборка De novo была выполнена с помощью Supernova v2.1.1 (ассемблер Supernova, RRID: SCR_016756) [44] с использованием предложенных параметров (—maxreads = 2100000000 —accept-extreme-охват —nopreflight).TGS-GapCloser v1.0.0 (TGS-GapCloser, RRID: SCR_017633) [45,46] использовался для заполнения пробелов между контигами внутри одних и тех же каркасов, и этот процесс был выполнен Canu с использованием исправленных ошибок ONT или данных PacBio . Количество зазоров внутри каркасов рассчитывали по формуле: количество контигов — количество каркасов.
Технические характеристики вычислительных кластеров, используемых в этом исследовании, представлены в дополнительной таблице S1. Оценка вычислительных затрат на основе цен на Amazon EC2 по требованию приведена в дополнительной таблице S2.
Оценка сборки
Статистика сборкибыла рассчитана с использованием QUAST v5.0.2 (QUAST, RRID: SCR_001228) [47] с минимальной длиной контига 10 КБ и параметрами —fragmented —large. Общедоступный эталонный геном M. integrifolia v2 (доступ в Genbank: GCA_
1585.1) [8] был использован в качестве эталонного генома для QUAST. Чтобы оценить базовую точность, QUAST использовался для вычисления количества несовпадений и отступов по сравнению со сборкой короткого чтения Illumina, созданной SPAdes.Сборка коротких считываний Illumina была создана с использованием более точных коротких считываний по сравнению с длинными считываниями; поэтому он содержал меньше базовых ошибок. Следовательно, количество несоответствий и отступов, выявленных в сборках для длительного чтения, по сравнению с сборкой для короткого чтения, будет отражать их базовую частоту ошибок. Мы отметили, что это позволит сравнить только X% генома, потому что сборка только для Illumina является относительно неполной. Кроме того, ожидается, что сборка Illumina будет иметь ошибки, и эти ошибки приведут к вызову ошибок в других сборках, даже если они действительно верны.Для оценки полноты генома сборки были подвергнуты BUSCO v3.0.2 (BUSCO, RRID: SCR_015008) [48] с базой данных eudicotyledons_odb10 (2121 ген). K-mer Analysis Toolkit v2.4.2 (KAT, RRID: SCR_016741) [49] Команды comp и kat_distanalysis использовались для оценки полноты сборки k -mer со ссылкой на короткие чтения Illumina или stLFR.
Результаты
Сборка генома Illumina
В результате секвенирования Illumina было получено 112.5 миллионов считываний парных концов длиной 150 пар оснований, что соответствует примерно 41-кратному охвату генома. После адаптера и обрезки хвоста polyG были собраны короткие считывания с использованием программного обеспечения SPAdes. Полученная в результате сборка состояла из 1 631 183 контигов общей длиной 864 Мб и содержала 15 583 контига> 10 кб с общей длиной 338 Мб (дополнительная таблица S3). Сборка была сильно фрагментирована, с контигом N50 размером 23,9 kb. Оценка полноты генома с помощью BUSCO показала, что сборка содержала 65% полных BUSCO (включая 58% однокопийных генов), 18% фрагментированных BUSCO и 17% отсутствующих BUSCO.
Сборка генома ONT
Для секвенирования ONT мы объединили результаты 1 проточной кюветы PromethION и 1 проточной кюветы MinION, получив в общей сложности 24,9 Гб данных с длиной чтения N50 27,8 кб (таблица 1). Проточная кювета PromethION и проточная кювета MinION генерировали 23,2 и 1,7 Гб данных соответственно с длиной считывания N50 28,5 и 16,6 кбайт и средним качеством считывания 6,3 и 8,9. Считывания ONT были собраны с использованием 4 различных ассемблеров длительного считывания (Redbean, Flye, Canu, Raven) и 3 разных подмножеств считывания, представляющих разный охват генома (21 ×, 28 × и 32 ×).Статистика для каждой сборки представлена в дополнительной таблице S4 и на рис. S1. Canu и Flye сгенерировали самые большие и наиболее смежные сборки, в то время как Redbean произвела самую маленькую и наименее смежную сборку (∼750 Mb, contig N50 ∼700 kb), за которой последовал Raven (∼770 Mb, contig N50 ∼1 Mb). Flye последовательно производил сборки размером ~ 812 Мб с контигом N50 ~ 1,5 Мб, тогда как смежность сборок Canu и Redbean увеличивалась по мере увеличения охвата считыванием. В частности, Canu contig N50 значительно увеличился с 706 kb (21x) до 1.43 Мб (32 ×). Для 28-кратного и 32-кратного покрытия генома сборки Raven были одинаковыми по размеру (Raven — единственный инструмент, для которого не требуется предполагаемый размер генома в качестве обязательного входного параметра). Raven был единственным инструментом, работающим на сервере с ускорением на GPU, и он был самым быстрым ассемблером, за ним шли Redbean и Flye. Canu был в 5 и 10 раз медленнее Flye и Redbean соответственно.
| Параметр . | ОНТ . | PacBio . | BGI . | Illumina . | |||||
|---|---|---|---|---|---|---|---|---|---|
| Количество необработанных считываний | 3,129,385 | 3,170,206 | 738,145,698 | 112,508,072 | |||||
| Кол-во отсеченных считываний 9125 | используется в сборке | 3,129,385 | 3,170,206 | 372,797,279 | 109,046,265 | ||||
| No.баз | 24,915,207,810 | 65,228,232,554 | 74,559,455,800 | 31,961,393,885 | |||||
| Длина считывания N50 (bp) | 27,842 | 35,866 9125 9125 9125 9125 | 35866 912 912 912 912 9125 9125 9125 | 2 | 7,962 | 20575 | 2 × 100 | 2 × 150 | |
| Покрытие генома (x) | 32 | 84 | 96 | 41 | |||||
| долларов США Стоимость (долл. США) | 12,560 | 1,120 | 721 | ||||||
| Дата установления последовательности | март / апрель 2019 г. | июнь 2019 г. | май / июнь 2019 г. | апрель 2019 г. | |||||
| Количество ДНК (нг58 | –1258 | –1258 | 9 15000 | 10 | 500 |
| Параметр . | ОНТ . | PacBio . | BGI . | Illumina . | |||||
|---|---|---|---|---|---|---|---|---|---|
| Количество необработанных считываний | 3,129,385 | 3,170,206 | 738,145,698 | 112,508,072 | |||||
| Кол-во отсеченных считываний 9125 | используется в сборке | 3,129,385 | 3,170,206 | 372,797,279 | 109,046,265 | ||||
| No.баз | 24,915,207,810 | 65,228,232,554 | 74,559,455,800 | 31,961,393,885 | |||||
| Длина считывания N50 (bp) | 27,842 | 35,866 9125 9125 9125 9125 | 35866 912 912 912 912 9125 9125 9125 | 2 | 7,962 | 20575 | 2 × 100 | 2 × 150 | |
| Покрытие генома (x) | 32 | 84 | 96 | 41 | |||||
| долларов США Стоимость (долл. США) | 12,560 | 1,120 | 721 | ||||||
| Дата установления последовательности | март / апрель 2019 г. | июнь 2019 г. | май / июнь 2019 г. | апрель 2019 г. | |||||
| Количество ДНК (нг58 | –1258 | –1258 | 9 15000 | 10 | 500 |
| Параметр . | ОНТ . | PacBio . | BGI . | Illumina . | |||||
|---|---|---|---|---|---|---|---|---|---|
| Количество необработанных считываний | 3,129,385 | 3,170,206 | 738,145,698 | 112,508,072 | |||||
| Кол-во отсеченных считываний 9125 | используется в сборке | 3,129,385 | 3,170,206 | 372,797,279 | 109,046,265 | ||||
| No.баз | 24,915,207,810 | 65,228,232,554 | 74,559,455,800 | 31,961,393,885 | |||||
| Длина считывания N50 (bp) | 27,842 | 35,866 9125 9125 9125 9125 | 35866 912 912 912 912 9125 9125 9125 | 2 | 7,962 | 20575 | 2 × 100 | 2 × 150 | |
| Покрытие генома (x) | 32 | 84 | 96 | 41 | |||||
| долларов США Стоимость (долл. США) | 12,560 | 1,120 | 721 | ||||||
| Дата установления последовательности | март / апрель 2019 г. | июнь 2019 г. | май / июнь 2019 г. | апрель 2019 г. | |||||
| Количество ДНК (нг58 | –1258 | –1258 | 9 15000 | 10 | 500 |
| Параметр . | ОНТ . | PacBio . | BGI . | Illumina . | ||||
|---|---|---|---|---|---|---|---|---|
| Количество необработанных считываний | 3,129,385 | 3,170,206 | 738,145,698 | 112,508,072 | ||||
| Кол-во отсеченных считываний 9125 | используется в сборке | 3,129,385 | 3,170,206 | 372,797,279 | 109,046,265 | |||
| No.баз | 24,915,207,810 | 65,228,232,554 | 74,559,455,800 | 31,961,393,885 | ||||
| Длина считывания N50 (bp) | 27,842 | 35,866 9125 9125 9125 9125 | 35866 912 912 912 912 9125 912 9125 912 9125 9125 9125 9125 9126 | 7,962 | 20575 | 2 × 100 | 2 × 150 | |
| Покрытие генома (x) | 32 | 84 | 96 | 41 | ||||
| долларов США Стоимость (долл. США) | 12,560 | 1,120 | 721 | |||||
| Дата установления последовательности | март / апрель 2019 г. | июнь 2019 г. | май / июнь 2019 г. | апрель 2019 г. | ||||
| Количество ДНК (нг58) 1,2001259 | 9 15,000 | 10 | 500 |
Впоследствии мы отполировали Redbean, Flye, Canu и Rav ru наброски сборок с использованием длинных чтений ONT, за которыми следуют короткие чтения Illumina.Долговременная полировка проводилась с помощью инструментов Racon и Medaka. Было проведено сравнение двух программных инструментов для исправления базовых ошибок с помощью коротких чтений: широко используемого инструмента Pilon и недавно разработанного алгоритма NextPolish. Эти этапы полировки значительно улучшили полноту генома, о чем свидетельствует процент полных BUSCO, который увеличился с 53% (Redbean), 70% (Canu) или 79% (Flye, Raven) до 85% (Redbean) или 89% ( Flye, Raven, Canu) после полировки с длинным считыванием и 92% (Redbean) или 95% (Flye, Raven, Canu) после полировки с длинным и коротким считыванием (дополнительная таблица S5 и рис.S2). В качестве оценки базовой точности мы вычислили количество несовпадений и отступов по сравнению с узлом короткого чтения Illumina, созданным SPAdes (дополнительный рисунок S3 и таблица S6). Сборка Canu была менее точной, чем другие сборки (NextPolish: 582 против 485–503 несоответствий на 100 кб, 68 против 42–49 инделей на 100 кб; Pilon: 670 против 529–593 несоответствий на 100 кб, 108 против 76–85 indels на 100 т.п.н.) и содержали более высокий процент дуплицированных генов (16–17% против 12–14%).
Базовые показатели точности показывают, что NextPolish работает немного лучше, чем Pilon.В частности, количество отступов было значительно уменьшено после полировки с помощью NextPolish по сравнению с Pilon (Flye: 48 против 83 отступов на 100 КБ, Canu: 68 против 108, Raven: 49 против 85, Redbean: 42 против 76, дополнительная таблица S6 ). Pilon и NextPolish привели к аналогичной полноте генома при применении к сборкам Canu и Raven. Полнота генома была немного лучше после 2 итераций NextPolish, чем после 2 итераций Pilon for the Flye (95,4% против 95,2%) и сборок Redbean (91,9% против 91.6%). Вторая итерация Pilon привела к небольшому уменьшению количества отсутствующих генов и более высокой точности для всех 4 ассемблеров, тогда как вторая итерация NextPolish не улучшила полноту и точность генома (несоответствия) для сборок Canu и Raven. Следовательно, в зависимости от сборщика и используемого полировщика количество рекомендуемых итераций полировки может быть разным.
Полнота сборки также оценивалась путем сравнения спектра k -мерных полированных сборок со спектром k -мерных коротких считываний Illumina (дополнительная таблица S7 и рис.S4). Анализ k -mer показал, что Flye произвел наиболее полную полированную сборку (99,0%), за ней следуют Canu (97,9%) и Raven (97,4%) и, наконец, Redbean (92,3%). Тенденции были аналогичными, когда анализ k -меров выполнялся с использованием коротких считываний stLFR.
В качестве метода, альтернативного сборке только для длительного чтения с последующей полировкой короткими считываниями, с помощью MaSuRCA была сгенерирована гибридная сборка. Сборка ONT + Illumina показала аналогичный размер (797 Мб), смежность (contig N50 = 1.18 Мб), полноту (94,8% завершенных BUSCO, включая 15,5% дублированных BUSCO) и немного более низкую точность (530 несоответствий на 100 кбайт, 53 отступа на 100 кбайт), как показывает сборка Flye и Raven с последующей полировкой с помощью Illumina (рис. –3 и дополнительный рис. S2 и S3, таблицы S4 и S5). Полировка коротким считыванием или длительное считывание с последующей полировкой короткого считывания существенно не улучшили полноту генома сборки MaSuRCA (дополнительная таблица S5), что ожидается, поскольку суперчитания, построенные этим инструментом, основаны на считываниях Illumina.
Рисунок 1. Статистика сборки генома
ONT, PacBio и BGI. Общая длина сборки отображается в зависимости от контига N50 для каждого набора данных ассемблера и секвенирования.
Рисунок 1:
Статистика сборки генома ONT, PacBio и BGI. Общая длина сборки отображается в зависимости от контига N50 для каждого набора данных ассемблера и секвенирования.
Рисунок 2:
Количество несоответствий и отступов, выявленных в сборках для длительного чтения, по сравнению с сборкой для короткого чтения Illumina, созданной SPAdes.Сборки BGI + ONT и BGI + PacBio были отполированы с помощью чтения BGI stLFR с использованием 1 итерации NextPolish. Сборки ONT + Illumina (кроме MaSuRCA) были отполированы с помощью длинных чтений ONT с использованием Racon и Medaka, за которыми следовали короткие чтения Illumina с использованием 1 итерации NextPolish. Сборки PacBio + Illumina (кроме MaSuRCA) были отполированы короткими считываниями Illumina с использованием 1 итерации NextPolish. * Сборка отполирована с помощью Illumina читает.
Рисунок 2:
Количество несоответствий и отступов, выявленных в сборках для длительного чтения, по сравнению с сборкой для короткого чтения Illumina, созданной SPAdes.Сборки BGI + ONT и BGI + PacBio были отполированы с помощью чтения BGI stLFR с использованием 1 итерации NextPolish. Сборки ONT + Illumina (кроме MaSuRCA) были отполированы с помощью длинных чтений ONT с использованием Racon и Medaka, за которыми следовали короткие чтения Illumina с использованием 1 итерации NextPolish. Сборки PacBio + Illumina (кроме MaSuRCA) были отполированы короткими считываниями Illumina с использованием 1 итерации NextPolish. * Сборка отполирована с помощью Illumina читает.
Рисунок 3:
BUSCO анализ сборок с использованием набора данных eudicotyledons (2121 ген).Ось X отображает процент полной и единственной копии, полной и дублированной, фрагментированной и отсутствующей BUSCO, а ось Y показывает оцениваемую сборку. Сборки BGI + ONT и BGI + PacBio были отполированы с помощью чтения BGI stLFR с использованием 1 итерации NextPolish. Сборки ONT + Illumina (кроме MaSuRCA) были отполированы с помощью длинных чтений ONT с использованием Racon и Medaka, за которыми следовали короткие чтения Illumina с использованием 1 итерации NextPolish. Сборки PacBio + Illumina (кроме MaSuRCA) были отполированы короткими считываниями Illumina с использованием 1 итерации NextPolish.
Рисунок 3:
Анализ сборок BUSCO с использованием набора данных eudicotyledons (2121 ген). Ось X отображает процент полной и единственной копии, полной и дублированной, фрагментированной и отсутствующей BUSCO, а ось Y показывает оцениваемую сборку. Сборки BGI + ONT и BGI + PacBio были отполированы с помощью чтения BGI stLFR с использованием 1 итерации NextPolish. Сборки ONT + Illumina (кроме MaSuRCA) были отполированы с помощью длинных чтений ONT с использованием Racon и Medaka, за которыми следовали короткие чтения Illumina с использованием 1 итерации NextPolish.Сборки PacBio + Illumina (кроме MaSuRCA) были отполированы короткими считываниями Illumina с использованием 1 итерации NextPolish.
Сборка генома PacBio
Используя 8 одномолекулярных ячеек реального времени на платформе PacBio Sequel, мы сгенерировали 3 170 206 субпотоков с длиной чтения N50, равной 35,9 кб, что в сумме составляет 65,2 ГБ (таблица 1). Данные соответствуют примерно 84-кратному охвату предполагаемого размера генома в 780 мегабайт. Сборка данных PacBio проводилась с использованием тех же инструментов, которые использовались для данных ONT: 4 ассемблера длительного чтения Redbean, Flye, Canu и Raven и гибридного ассемблера MaSuRCA (дополнительная таблица S8).Сборки PacBio демонстрировали такую же смежность, что и сборки ONT (кроме Canu), и были больше (за исключением Flye) (рис. 1). Перед полировкой их полнота генома была выше, чем у сборок ONT, что указывает на более высокую точность считывания PacBio (дополнительный рис. S2). Сборка Redbean была наиболее фрагментированной (contig N50 = 649 kb) и наименее полной (89% полных BUSCO). Сборка Flye была очень смежной (contig N50 = 1,47 Мб) и была самой маленькой по размеру (767 Мб). Сборка Raven (879 МБ) состояла из наименьшего количества контигов (n = 1730) с контигом N50 919 kb.Сборка Canu была самой большой (1,2 ГБ), но она содержала высокую долю дупликации, как сообщает QUAST (1,64) и подтверждается процентом дублированных BUSCO (53%) и спектрами k -меров (дополнительный рис. ). Следовательно, сборка Canu, вероятно, содержит несколлапсированные гаплотипы, соответствующие артефактически дублированным регионам, как недавно сообщалось [50]. Выравнивание сборок PacBio со сборкой M. integrifolia выявило большее количество неправильных сборок в сборке Canu (n = 38 800) по сравнению с другими сборками (n = 21 000–27 000).Гибридная сборка PacBio + Illumina (807 МБ, contig N50 = 1,22 МБ) содержала 94,9% полных BUSCO, включая 16% дуплицированных генов (рис. 3).
Для создания поэтапной диплоидной сборки, затем была проведена сборка PacBio с использованием ассемблера FALCON с последующим разрешением гаплотипов и полировкой с помощью FALCON-Unzip. Полученная первичная сборка состояла из 1333 контигов общей длиной 871 МБ, при этом половина сборки состояла из контигов размером 1,38 МБ или более (рис. 1). FALCON-Unzip также сгенерировал 2488 альтернативных гаплотигов размером 495 Мб (т.е.е. 57% генома было разрешено по гаплотипу) с контигом N50 в 333 т.п.н. Анализ BUSCO на первичных контигах показал ~ 26% дуплицированных генов, что позволяет предположить наличие гомологичных первичных контигов (рис. 3). Конвейер Purge Haplotigs идентифицировал 569 первичных контигов, представляющих 112 МБ, как вероятные альтернативные гаплотипы (дополнительная таблица S9). Эти контиги были перенесены в набор гаплотигов. Курируемая первичная гаплоидная сборка состояла из 762 контигов общим размером 758 Мб с контигом N50 1,59 Мб и содержала меньше дублированных генов (16%) с минимальным влиянием на полноту генома (полные BUSCO на 95%).
Впоследствии мы отполировали сборки PacBio, используя короткие чтения Illumina. Как и ожидалось, в сборках было выявлено меньшее количество несовпадений и дефектов по сравнению со сборкой Illumina (дополнительный рисунок S3 и таблица S6). Полировка уменьшает количество отсутствующих BUSCO, но увеличивает количество дублированных BUSCO для сборок Redbean, Flye и Raven (дополнительная таблица S10). Долгое считывание с последующей полировкой короткого чтения привело к увеличению процента однокопийных BUSCO и уменьшенному проценту дублированных BUSCO для сборки Canu и, в меньшей степени, сборки Falcon.Интересно, что этап полировки с длинным считыванием не улучшил полноту сборки Redbean, Flye и Raven, и аналогичные или немного лучшие результаты были получены только после полировки с коротким считыванием. Следовательно, рекомендуемая стратегия полировки сборок PacBio может зависеть от используемого ассемблера.
Использование отфильтрованного по качеству подмножества субпотоков (эквивалентного 67-кратному охвату генома) привело к аналогичной (Flye и Raven) или немного более высокой (Redbean) смежности сборки, не влияя на полноту генома (только Redbean, Raven и Flye). были протестированы из-за высоких вычислительных требований Canu и Falcon) (дополнительный рис.S1 и Таблица S8). Наконец, чтобы сравнить технологии PacBio и ONT, мы случайным образом выполнили субдискретизацию субпотоков PacBio до покрытия, эквивалентного данным ONT (32 ×). Полученная в результате сборка Flye показала аналогичный размер 764 МБ, более низкую смежность (contig N50 = 1,26 МБ) и такую же полноту генома (94,7% полных BUSCO), что и сборка покрытия 84x (рис.1, дополнительный S2 и таблица S8). ). Остальные 4 ассемблера привели к уменьшению размера генома и немного меньшей полноте генома. Уменьшение покрытия не повлияло на смежность сборки Raven (contig N50 = 894 kb).Сборка Falcon больше всего пострадала от уменьшения покрытия, при уменьшении contig N50 с 1,38 Мб до 684 Кб. Напротив, смежность сборки Redbean увеличилась с 649 до 953 kb. Процент дублированных BUSCO уменьшился для всех сборок, но остался высоким для сборок Canu (33%) и Falcon (20%).
Сборка генома stLFR
stLFR сгенерировал 738 миллионов операций чтения на парном конце длиной 100 пар оснований. Чтобы соответствовать требованиям ассемблера, штрих-коды с <10 считываний были удалены, в результате чего было выполнено 373 миллиона считываний, что составляет 74.6 Гб данных и соответствует ∼96-кратному охвату генома (Таблица 1). Считывания stLFR были собраны с использованием Supernova2 в сборку из 40 789 каркасов общей длиной 880 МБ (таблица S11). Всего 5065 каркасов были больше 10 т.п.н., с общей длиной 752 МБ и N50 3,54 МБ для каркаса и 35,6 т.п.н. для контига (таблица 2). По сравнению с сборкой коротких считываний Illumina сборка stLFR содержала наименьшее количество несовпадений и отступов (рис. 2). Анализ консервативного гена BUSCO показал, что сборка stLFR содержит 88.3% полных генов из набора данных eudicotyledons (рис. 3).
Таблица 2:Заполнение зазора для сборки stLFR с использованием исправленного ONT или PacBio читает
| Параметр . | Сверхновая . | ОНТ . | PacBio . | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| После заполнения зазора . | Улучшение (%) . | После заполнения зазора . | Улучшение (%) . | ||||||||
| Число вводимых длинных считываний | 1,056,095 | 674,796 | |||||||||
| Используемых считываний для заполнения (%) | 58 | Кол-во подмостей | 5,065 | 5,332 | 5,3 | 5,446 | 7,5 | ||||
| Подмостей N50 (п.5 | 3,504,721 | −1,0 | |||||||||
| Длина каркаса (bp) | 751,745,340 | 766,968,089 | 2,0 | 768,468,395 | ,48 8 | Размер наибольшего 3,3 | 31,237,530 | 3,6 | |||
| Число контигов | 19,954 | 6,022 | −70 | 5,717 | −71 | ||||||
| p | 1,598,608 | 4,390 | |||||||||
| Длина контига (п. | 23 824 472 | 4 499 | 91 254|||||||||
| No.зазоров внутри каркасов | 14,889 | 690 | −95 | 271 | −98 | ||||||
| Кол-во н. | |||||||||||
| Число завершенных BUSCO (%) | |||||||||||
| Все | 1873 (88,3) | 1,963 (92,5) | 4,8 | 5)5,8 | |||||||
| Однократная | 1,646 (77,6) | 1,710 (80,6) | 3 | 1,679 (79,2) | 1,6 | ||||||
| Дублированная | 2259 (11,9) | 1,2 | 304 (14,3) | 3,6 | |||||||
| Параметр . | Сверхновая . | ОНТ . | PacBio . | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| После заполнения зазора . | Улучшение (%) . | После заполнения зазора . | Улучшение (%) . | ||||||||
| Число вводимых длинных считываний | 1,056,095 | 674,796 | |||||||||
| Используемые чтения для заполнения (%) | 59 | №подмостей | 5,065 | 5,332 | 5,3 | 5,446 | 7,5 | ||||
| Подмостей N50 (bp) | 3,540,919 | 3,23,921 | 5длина п.3 | 31,237,530 | 3,6 | ||||||
| Кол-во контигов | 19,954 | 6,022 | -70 | 5,717 | −71 | ||||||
| p | 46 | 46 | 1,598,608 | 4,390 | |||||||
| Длина контига (п. | 23,824,472 | 4,499 | |||||||||
| №зазоров внутри каркасов | 14,889 | 690 | −95 | 271 | −98 | ||||||
| Кол-во н. | |||||||||||
| Число завершенных BUSCO (%) | |||||||||||
| Все | 1873 (88,3) | 1,963 (92,5) | 4,8 | 5)5,8 | |||||||
| Однократная | 1,646 (77,6) | 1,710 (80,6) | 3 | 1,679 (79,2) | 1,6 | ||||||
| Дублированная | 2259 | 2259 (11,9) | 1,2 | 304 (14,3) | 3,6 | ||||||
Заполнение зазора для сборки stLFR с использованием исправленного ошибок ONT или PacBio считывает
| Параметр . | Сверхновая . | ОНТ . | PacBio . | |||
|---|---|---|---|---|---|---|
| После заполнения зазора . | Улучшение (%) . | После заполнения зазора . | Улучшение (%) . | |||
| Количество вводимых длинных считываний | 1,056,095 | 674,796 | ||||
| Полезные чтения для заполнения (%) | 1.74 | 2,95 | ||||
| Кол-во подмостей | 5,065 | 5,332 | 5,3 | 5,446 | 7,5 | |
| 3,504,721 | −1,0 | |||||
| Длина подмостей (bp) | 751,745,340 | 766,968,089 | 2,0 | 768,468,395 | 2.2 | |
| Наибольший размер каркаса (п. 71 | ||||||
| Contig N50 (bp) | 35,605 | 1,046,570 | 2,839 | 1,598,608 | 4,390 | |
| 98 9125 | 98 | |||||
| Наибольший размер контига (п.зазоров внутри каркасов | 14,889 | 690 | −95 | 271 | −98 | |
| Кол-во н. | ||||||
| Число завершенных BUSCO (%) | ||||||
| Все | 1873 (88,3) | 1,963 (92,5) | 4,8 | 5)5,8 | ||
| Однократная | 1,646 (77,6) | 1,710 (80,6) | 3 | 1,679 (79,2) | 1,6 | |
| Дублированная | 2259 (11,9) | 1,2 | 304 (14,3) | 3,6 | ||
| Параметр . | Сверхновая . | ОНТ . | PacBio . | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| После заполнения зазора . | Улучшение (%) . | После заполнения зазора . | Улучшение (%) . | ||||||||
| Число вводимых длинных считываний | 1,056,095 | 674,796 | |||||||||
| Используемые чтения для заполнения (%) | 59 | №подмостей | 5,065 | 5,332 | 5,3 | 5,446 | 7,5 | ||||
| Подмостей N50 (bp) | 3,540,919 | 3,23,921 | 5длина п.3 | 31,237,530 | 3,6 | ||||||
| Кол-во контигов | 19,954 | 6,022 | -70 | 5,717 | −71 | ||||||
| p | 46 | 46 | 1,598,608 | 4,390 | |||||||
| Длина контига (п. | 23,824,472 | 4,499 | |||||||||
| №зазоров внутри каркасов | 14,889 | 690 | −95 | 271 | −98 | ||||||
| Кол-во н. | |||||||||||
| Число завершенных BUSCO (%) | |||||||||||
| Все | 1873 (88,3) | 1,963 (92,5) | 4,8 | 5)5,8 | |||||||
| Однократная | 1,646 (77,6) | 1,710 (80,6) | 3 | 1,679 (79,2) | 1,6 | ||||||
| Дублированная | 2259 (11.9) | 1,2 | 304 (14,3) | 3,6 | |||||||
Включение данных ONT или PacBio для заполнения пробелов в каркасах привело к 29- или 45-кратному увеличению длины контига N50 с 35,6 кб. к 1.05 или 1,60 мб, а также уменьшение в 22 или 55 раз количества пробелов внутри каркасов размером более 10 т.п.н. с 14 889 до 690 или 271 (таблица 2). Подмость N50 незначительно уменьшилась на 0,02 или 0,04 Мб за счет корректировки предполагаемых зазоров. Для обеих сборок с заполнением зазоров общая длина сборки увеличилась соответственно до ~ 895 и 770 мб для каркасов размером более 10 тп. Размер наибольшего контига увеличился с 518 Кб до 9,7 Мб (ONT) и 23,8 Мб (PacBio). Кроме того, полнота генома была улучшена в сборках, заполненных пробелами, с помощью BUSCO, обнаруживающего 4.Более полные гены на 8% (ONT) и на 5,8% (PacBio). Количество полностью дублированных BUSCO было немного ниже в сборке с заполнением ONT (11,9%), чем в сборке с заполнением PacBio (14,3%). Наконец, расчетная полнота сборки k -меров увеличилась в сборках с зазором с 95,8% до 96,7% (ONT) и 97,4% (PacBio) (дополнительная таблица S7). Дальнейшая полировка сборок с заполненными промежутками с использованием считываний stLFR привела к небольшому увеличению полноты генома до 93,2% (ONT) и 93.7% (PacBio) полных генов BUSCO (дополнительная таблица S11 и рис. S2) и уменьшение количества инделей (дополнительная таблица S6 и рис. S3).
Обсуждение
Мы сообщаем о сравнении трех наборов данных длительного чтения, полученных из одного и того же образца ДНК растений. M. jansenii был выбран для этого исследования из-за его важности для сохранения и разведения. Все 4 вида Macadamia перечислены как находящиеся под угрозой исчезновения согласно австралийскому законодательству, но M.jansenii особенно уязвим, поскольку он был зарегистрирован только в одном месте. M. jansenii не был одомашнен, и его мелкие горькие орехи являются препятствием, ограничивающим простое вмешательство в процесс размножения. Однако характерный небольшой размер дерева, который на 50% меньше, чем у коммерческих сортов, представляет интерес для использования при проектировании садов с высокой плотностью посадки, и для этой цели они проходят испытания в качестве подвоя [51]. Это самый северный вид Macadamia и может быть источником генов адаптации к более теплому климату [52].Получены гибриды M. integrifolia и M. jansenii .
Три технологии секвенирования с длинным чтением значительно улучшили полноту сборки по сравнению со сборкой, произведенной с использованием только чтения Illumina (65% полных BUSCO). Стоимость создания 1 Гб данных секвенирования (включая подготовку библиотеки) составила 193 доллара США для PacBio Sequel I, 97 долларов США для ONT PromethION и 12 долларов США для BGI stLFR (необработанные считывания, впоследствии используемые при сборке). Виртуальные длинные чтения были сгенерированы с использованием протокола stLFR.Эта технология извлекает выгоду из точности и низкой стоимости платформы короткого считывания секвенирования, обеспечивая при этом информацию на большом расстоянии. stLFR был самым дешевым подходом, и он генерировал сборку с наименьшим количеством ошибок single base и indel. Кроме того, сборка, произведенная Supernova, была поэтапной. При этом сборка stLFR была более фрагментирована, чем другие технологии длительного чтения. Мы также продемонстрировали, что stLFR можно использовать в качестве дополнительной технологии к ONT. В самом деле, включение прочтений Nanopore значительно увеличило смежность сборки stlFR, при этом N50 достигло 1 Мб, и улучшило полноту генома.Интересно, что на этапе заполнения пробелов использовалось только 1,7% считываний ONT, предполагая, что подход селективного секвенирования в реальном времени может быть использован для выбора конкретных молекул, которые будут информативными для заполнения пробелов [53].
Когда все чтения были включены, сборки, созданные с использованием данных PacBio и ONT, были сопоставимы с точки зрения смежности сборки (contig N50 ~ 1,5 Мб) и полноты генома (95% полных BUSCO). Однако, когда мы использовали одинаковый объем данных для каждой платформы (32-кратное покрытие), смежность сборки PacBio, произведенной Falcon, уменьшилась вдвое и стала только вдвое меньше, чем у сборок ONT Flye или Canu.Ассемблеры Flye и Raven оказались более устойчивыми к падению покрытия PacBio, поскольку контиг сборки N50 уменьшился только с 1,47 до 1,26 Мбайт (Flye) и с 919 до 894 Кбайт (Raven). Кроме того, мы обнаружили, что полировка сборки ONT с помощью коротких считываний Illumina требовалась для достижения полноты генома, аналогичной полноте сборки PacBio. Как для данных ONT, так и для PacBio, наивысшая смежность была получена с полированной сборкой с длинным считыванием по сравнению с гибридной сборкой, включающей как короткие, так и длинные считывания.
С тех пор, как были сгенерированы данные о последовательностях, платформа PacBio SMRT перешла с Sequel I на Sequel II с 8-кратным увеличением объема данных. Последняя платформа обеспечивает высокоточные чтения, которые более точны, чем непрерывные длинные чтения, собранные в этом исследовании. Следовательно, стоимость создания аналогичной сборки PacBio в системе Sequel II будет значительно снижена, а качество сборки, вероятно, улучшится, требуя меньше вычислительных ресурсов.
Требования к материалу ДНК для подготовки библиотеки секвенирования — еще один важный параметр, который следует учитывать при выборе технологии секвенирования. Для секвенирования ONT рекомендуется получить ≥1–2 мкг высокомолекулярной ДНК. Для создания библиотеки stLFR требуется ≥10 нг высокомолекулярной ДНК. Для секвенирования PacBio SMRT требуется высокий ввод геномной ДНК — 5–20 мкг высокомолекулярной ДНК для стандартного протокола библиотеки в зависимости от размера генома, но протокол низкого ввода ДНК PacBio снизил это требование до 100 нг на 1 ГБ генома. размер [54].Кроме того, PacBio недавно выпустила основанный на амплификации протокол ввода сверхнизкой ДНК, начиная с 5 нг высокомолекулярной ДНК.
Необходимо учитывать вычислительные требования и связанные с ними затраты, и они будут в значительной степени зависеть от размера генома интересующего вида. Были важные различия во времени выполнения сборки и использовании памяти в зависимости от используемого инструмента. Например, полировка с коротким чтением с использованием NextPolish использовала меньше памяти, чем Pilon, при этом обеспечивая аналогичные результаты.Вычисления с ускорением на GPU значительно сократили время вычислений для некоторых инструментов, таких как Racon, Medaka или Raven. Есть также проблемы, связанные с быстрым развитием технологий и программного обеспечения. Например, мы наблюдали значительное улучшение непрерывности сборки ONT в зависимости от используемой версии basecaller или ассемблера. В новейших выпусках ассемблеров, таких как Canu v2.1, Flye v2.8 или Raven v1.1.10, вероятно, будут созданы улучшенные сборки.
Три технологии длительного чтения позволили получить очень смежные и полные сборки генома.Затем необходимы подходы к созданию каркасов дальнего действия, такие как захват конформации хромосом (Hi-C, Chicago) или технологии физических карт (оптическая карта, рестрикционная карта), чтобы упорядочить и ориентировать собранные контиги в каркасы длиной хромосомы [55].
Доступность данных
Данные секвенирования BGI, PacBio, ONT и Illumina, полученные в этом исследовании, были депонированы в SRA в рамках BioProject PRJNA609013 и BioSample SAMN14217788. Номера доступа следующие: BGI (SRR111
Дополнительные файлы
Таблица S1: Технические характеристики вычислительных кластеров
Таблица S2: Оценка вычислительных затрат на основе цен Amazon EC2 по требованию на 19 сентября 2020 г.
Таблица S3: Статистика сборки генома Illumina с использованием ассемблера SPAdes
Таблица S4: Статистика сборки генома ONT с использованием ассемблеров Redbean, Flye, Canu, Raven и MaSuRCA
Таблица S5: Оценка полноты генома BUSCO сборок ONT для длительного чтения (Redbean, Flye, Canu, Raven) и гибридных сборка (MaSuRCA)
Таблица S6: Статистика сборки QUAST с использованием сборки короткого чтения Illumina в качестве эталонного генома
Таблица S7: k -мерная полнота сборок ONT, PacBio и stLFR
Таблица S8: Статистика сборки генома PacBio с использованием ассемблеров Redbean, Flye, Falcon, Canu, Raven и MaSuRCA
Таблица S9: P Статистика сборки генома acBio и оценка полноты генома до и после Purge Haplotigs
Таблица S10: Оценка полноты генома BUSCO для сборок длительного считывания PacBio (Redbean, Flye, Falcon, Canu, Raven) и гибридной сборки (MaSuRCA)
Таблица S11: Статистика сборки генома BGI stLFR с использованием ассемблера Supernova и программного обеспечения для закрытия пробелов TGS-GapCloser
Рисунок S1: Статистика сборки генома.Общая длина сборки отображается в зависимости от контига N50 для каждого ассемблера и секвенирования покрытия. (A) сборки ONT, (B) сборки PacBio.
Рисунок S2: Оценка полноты генома BUSCO. (A) Сборки ONT до и после полировки с коротким считыванием Illumina с использованием 1 итерации NextPolish (Flye, Canu, Raven, Redbean) и гибридной сборки MaSuRCA, (B) сборки PacBio с использованием 32-кратного или 84-кратного покрытия секвенирования, (C) BGI Сборки stLFR до и после заполнения пробелов с использованием данных ONT или PacBio и после полировки с использованием чтения stLFR и 1 итерации NextPolish.
Рисунок S3: Количество несоответствий и отступов, выявленных в сборках для длительного чтения, по сравнению с сборкой для короткого чтения Illumina, созданной SPAdes. (A) Сборки ONT до и после полировки с коротким считыванием Illumina с использованием 1 итерации NextPolish (Flye, Canu, Raven, Redbean) и гибридной сборки MaSuRCA; (B) сборки PacBio до и после полировки с коротким считыванием Illumina с использованием 1 итерации NextPolish (Falcon, Flye, Canu, Raven, Redbean) и гибридной сборки MaSuRCA; (C) Сборки BGI stLFR до и после заполнения пробелов с использованием данных ONT или PacBio и после полировки с помощью чтения stLFR с использованием 1 итерации NextPolish.
Рисунок S4: Графики спектров k -меров из набора инструментов анализа k -меров, сравнивающие k -меры, обнаруженные в Illumina, с показаниями k -меров, обнаруженных в ONT, PacBio, stLFR и Сборки Illumina.
Сокращения
AUD: австралийские доллары; bp: пары оснований; BGI: Пекинский институт геномики; BUSCO: Бенчмаркинг универсальных ортологов единственной копии; BWA: выравниватель Берроуза-Уиллера; DNB: ДНК-наночастицы; дцДНК: двухцепочечная ДНК; Gb: пары гигабаз; гДНК: геномная ДНК; GPU: графический процессор; kb: пары килобаз; Mb: пары мегабаз; ONT: Oxford Nanopore Technologies; PacBio: Pacific Biosciences; QUAST: Инструмент оценки качества; SMRT: одиночная молекула в реальном времени; SPAdes: St.Петербургский сборщик генома; SRA: архив чтения последовательности; SRE: устранитель короткого чтения; stLFR: считывание длинных фрагментов в одной пробирке; SQB: буфер секвенирования; USD: доллар США.
Конкурирующие интересы
Сотрудники BGI (W.T., I.H., Q.Y., B.Y., O.W., M.X, P.W.), MGI (H.W.) и Complete Genomics (E.A., Q.M., R.D., B.A.P.) владеют пакетами акций BGI. Авторы заявляют, что у них нет других конкурирующих интересов.
Финансирование
Эта работа финансировалась Центром инноваций в геноме, Управлением исследовательской инфраструктуры, Квинслендский университет.Эта работа была частично поддержана планом Shenzhen Peacock Plan (NO.KQTD20150330171505310). L.J.M.C. был поддержан проектом Discovery с грантом DP170102626, присужденным Австралийским исследовательским советом.
Вклад авторов
А.Ф. подготовил образец. Б.Т. контролируемый сбор растений. С.К.Р. выполнили подготовку и секвенирование библиотеки ONT. T.J.C.B. выполнили подготовку библиотеки PacBio и секвенирование. В.М. выполняла сборки и оценку сборок Illumina, ONT и PacBio.Q.Y. и Х. выполнили подготовку и секвенирование библиотеки stLFR. I.H. контролировал и проверял подготовку и секвенирование библиотеки stLFR. W.T. выполнил сборку stLFR, заполнение пробелов и статистику для stLFR. E.A., Q.M., R.D., O.W. и B.A.P. разработали эксперименты по stLFR и провели анализ stLFR. M.X. и П. поддерживаются анализы stLFR. ОТ. просмотрел рукопись. В.М. написал рукопись при участии всех авторов. R.J.H. и L.J.M.C. разработал и курировал проект.
БЛАГОДАРНОСТИ
Мы благодарим Дуга Стетнера и Тома Каддихи за помощь с программным обеспечением Falcon, Николаса Роудса и Ченси Чжоу за помощь с программным обеспечением MaSuRCA, Таню Дуарте за обработку образца ДНК в гобелене, Мобашвер Алам за предоставление образцов ткани макадамии, Джоанну Кроуфорд и Линн Финк за поддержку в инициировании проекта и Сон Хоанг Нгуен за помощь в сборке каркасов генома.
Список литературы
1.Брутто
C
,Weston
P
.Macadamia jansenii (Proteaceae), новый вид из центрального Квинсленда
.Aust Syst Бот
.1992
;5
(6
):725
—8
. 3.Чейз
МВт
.Взаимоотношения семейств цветковых растений
. В:Генри
RJ
, изд.,Разнообразие и эволюция растений: генотипические и фенотипические вариации высших растений
.Уоллингфорд, Великобритания; Кембридж, Массачусетс
:CABI
;2005
.4.Brozynska
M
,Furtado
A
,Henry
RJ
.Геномика диких сородичей сельскохозяйственных культур: расширение генофонда для улучшения сельскохозяйственных культур
.Завод Биотехнология J
.2016
;14
(4
):1070
—85
.5.Abberton
M
,Batley
J
,Bentley
A
и др.Глобальная интенсификация сельского хозяйства в условиях изменения климата: роль геномики
.Завод Биотехнология J
.2016
;14
(4
):1095
—8
.6.Генри
RJ
.Инновации в генетике растений, адаптирующие сельское хозяйство к изменению климата
.Курр Опин Завод Биол
.2020
;56
:168
—73
.7.Niu
YF
,Li
GH
,Ni
SB
и др.Сборка генома и аннотация Macadamia tetraphylla
.2020
, DOI: .8.Nock
CJ
,Baten
A
,Mauleon
R
и др.Сборка в масштабе хромосом и аннотация генома макадамии ( Macadamia integrifolia HAES 741)
.G3 (Bethesda)
.2020
;10
(10
):3497
—504
. 9.Nock
CJ
,Baten
A
,Barkla
BJ
и др.Секвенирование генома и транскриптома характеризует пространство генов Macadamia integrifolia (Proteaceae)
.BMC Genomics
.2016
;17
(1
):937
.10.Пааянен
P
,Кеттлборо
G
,Лопес-Жирона
E
и др.Критическое сравнение технологий для проекта секвенирования генома растений
.Gigascience
.2019
;8
(3
), DOI: .11.Belser
C
,Istace
B
,Denis
E
и др.Хромосомные сборки геномов растений с использованием длинных считываний нанопор и оптических карт
.Нат Растения
.2018
;4
(11
):879
—87
.12.Logsdon
GA
,Vollger
MR
,Eichler
EE
.Секвенирование генома человека с длительным считыванием и его приложения
.Нат Рев Генет
.2020
;21
(10
):597
—614
. 13.Юнг
H
,Winefield
C
,Bombarely
A
и др.Инструменты и стратегии для секвенирования с длинным считыванием и сборки de novo геномов растений
.Trends Plant Sci
.2019
;24
(8
):700
—24
. 14.Wang
O
,Chin
R
,Cheng
X
и др.Эффективное и уникальное кобар-кодирование считываний секвенирования второго поколения с длинных молекул ДНК, обеспечивающее экономичное и точное секвенирование, гаплотипирование и сборку de novo
.Genome Res
.2019
;29
(5
):798
—808
.15.Drmanac
R
, изобретатель.Анализ нуклеиновых кислот случайными смесями неперекрывающихся фрагментов
. (13 июня,, 2006,
) 16.Peters
BA
,Liu
J
,Drmanac
R
.Последовательность с совместным штрих-кодом считывает длинные фрагменты ДНК: экономичное решение для секвенирования «идеального генома»
.Передний Genet
.2014
;5
:466
.17.Фуртадо
А
.Извлечение ДНК из вегетативной ткани для секвенирования нового поколения
.Методы Мол Биол
.2014
;1099
:1
—5
. 18.Wang
L
,Xi
Y
,Zhang
W
и др.Лигирование 3’-ответвлений: новый метод лигирования некомплементарной ДНК с углубленными или внутренними 3’OH-концами в ДНК или РНК
.ДНК Res
.2019
;26
(1
):45
—53
. 19.De Coster
W
,D’Hert
S
,Schultz
DT
и др.NanoPack: визуализация и обработка длинночитаемых данных секвенирования
.Биоинформатика
.2018
;34
(15
):2666
—9
.20.Фитиль
R
.Porechop: переходной триммер для Oxford Nanopore показывает
. .21.Фитиль
R
.Filtlong: качественный инструмент для фильтрации длинных чтений
. 23.Bolger
AM
,Lohse
M
,Usadel
B
.Trimmomatic: гибкий триммер для данных последовательности Illumina
.Биоинформатика
.2014
;30
(15
):2114
—20
.24.Chen
S
,Zhou
Y
,Chen
Y
и др.fastp: сверхбыстрый универсальный препроцессор FASTQ
.Биоинформатика
.2018
;34
(17
):i884
—90
. 25.Marçais
G
,Kingsford
C
.Быстрый подход без блокировок для эффективного параллельного подсчета появления k-мер
.Биоинформатика
.2011
;27
(6
):764
—70
. 26.Vurture
GW
,Sedlazeck
FJ
,Nattestad
M
и др.GenomeScope: быстрое безреференсное профилирование генома на основе коротких считываний
.Биоинформатика
.2017
;33
(14
):2202
—4
. 27.Руан
Дж
,Ли
H
.Быстрая и точная сборка с долгим чтением с wtdbg2
.Нат Методы
.2020
;17
(2
):155
—8
. 28.Колмогоров
M
,Юань
J
,Lin
Y
и др.Сборка длинных подверженных ошибкам операций чтения с использованием повторяющихся графиков
.Нат Биотехнология
.2019
;37
(5
):540
—6
.29.Koren
S
,Walenz
BP
,Berlin
K
, et al.Canu: масштабируемая и точная сборка с длинным считыванием за счет адаптивного взвешивания k-mer и разделения повторов
.Genome Res
.2017
;27
(5
):722
—36
. 30.Vaser
R
,Šikić
M
.Raven: de novo ассемблер генома для длинных чтений
.2020
, DOI: .31.Шанье
D
.Секвенирование полного генома видов плодовых деревьев
.Adv Bot Res
.2015
;74
, DOI: .32.Vaser
R
,Sović
I
,Nagarajan
N
и др.Быстрая и точная сборка генома de novo из длинных неисправленных считываний
.Genome Res
.2017
;27
(5
):737
—46
.33.Li
H
.Minimap2: попарное выравнивание нуклеотидных последовательностей
.Биоинформатика
.2018
;34
(18
):3094
—100
,35.Walker
BJ
,Abeel
T
,Shea
T
и др.Pilon: интегрированный инструмент для комплексного обнаружения вариантов микробов и улучшения сборки генома
.PLoS One
.2014
;9
(11
):e112963
,36.Li
H
.Считывает выравнивающую последовательность, последовательности клонирования и контиги сборки с BWA-MEM
.2013
: 1303.3997.37.Hu
J
,Вентилятор
J
,Sun
Z
и др.NextPolish: быстрый и эффективный инструмент для полировки генома для сборки с длинным считыванием
.Биоинформатика
.2020
;36
(7
):2253
—55
. 38.Зимин
AV
,Marçais
G
,Puiu
D
и др.Ассемблер генома MaSuRCA
.Биоинформатика
.2013
;29
(21
):2669
—77
. 39.Подбородок
CS
,Peluso
P
,Sedlazeck
FJ
и др.Фазовая диплоидная сборка генома с секвенированием одной молекулы в реальном времени
.Нат Методы
.2016
;13
(12
):1050
—4
.40.Roach
MJ
,Schmidt
SA
,Borneman
AR
.Purge Haplotigs: переназначение аллельного контига для диплоидных геномных сборок третьего поколения
.BMC Bioinformatics
.2018
;19
(1
):460
.41.Банкевич
А
,Нурк
S
,Антипов
D
и др.SPAdes: новый алгоритм сборки генома и его приложения для секвенирования отдельных клеток
.Дж. Comput Biol
.2012
;19
(5
):455
—77
. 43.Мартин
М
.Cutadapt удаляет последовательности адаптеров из операций чтения с высокой пропускной способностью
.EMBnet J
.2011
;17
(1
):10
. 44.Weisenfeld
NI
,Kumar
V
,Shah
P
и др.Прямое определение диплоидных последовательностей генома
.Genome Res
.2017
;27
(5
):757
—67
. 45.Xu
M
,Guo
L
,Gu
S
и др.TGS-GapCloser: быстрое и точное устройство закрытия пробелов для больших геномов с низким охватом подверженных ошибкам длинных считываний
.Gigascience
.2020
;9
(9
):giaa094
. 47.Гуревич
А
,Савельев
В
,Вяххи
N
и др.QUAST: Инструмент оценки качества сборки генома
.Биоинформатика
.2013
;29
(8
):1072
—5
.48.Simão
FA
,Waterhouse
RM
,Ioannidis
P
, et al.BUSCO: оценка сборки генома и полноты аннотации с однокопийными ортологами
.Биоинформатика
.2015
;31
(19
):3210
—2
. 49.Mapleson
D
,Garcia Accinelli
G
,Kettleborough
G
и др.KAT: K-mer Analysis Toolkit для контроля качества наборов данных NGS и геномных сборок
.Биоинформатика
.2017
;33
(4
):574
—6
,50.Guiglielmoni
N
,Derzelle
A
,van Doninck
K
и др.Преодоление несколлапсированных гаплотипов в долго читаемых сборках немодельных организмов
.2020
, DOI :.51.Alam
MM
,Wilkie
J
,Topp
BL
.Ранний рост и успешная прививка рассады макадамии и черенков
.Acta Hortic
.2018
; (1205
):637
—44
,52.Topp
BL
,Nock
CJ
,Hardner
CM
и др.Макадамия ( Macadamia spp.) разведение
. In:Al-Khayri
JM
,Jain
SM
,Johnson
DV
ред.Достижения в стратегиях селекции растений: ореховые культуры и культуры для производства напитков
.Cham
:Springer
;2019
:221
—51
. 53.Loose
M
,Malla
S
,Stout
M
.Селективное секвенирование в реальном времени с использованием нанопор
.Нат Методы
.2016
;13
(9
):751
—4
. 54.Kingan
S
,Heaton
H
,Cudini
J
и др.Сборка высококачественного генома de novo одного комара с использованием секвенирования PacBio
.Гены
.2019
;10
(1
):62
.55.Ghurye
J
,Pop
M
.Современные технологии и алгоритмы построения каркасов собранных геномов
.PLoS Comput Biol
.2019
;15
(6
):e1006994
.56.Murigneux
V
,Rai
SK
,Furtado
A
и др.Подтверждающие данные для «Сравнение методов длительного считывания для секвенирования и сборки генома растений».
.2020
. .© Автор (ы) 2020.Опубликовано издательством Oxford University Press GigaScience.
Это статья в открытом доступе, распространяемая в соответствии с условиями лицензии Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0/), которая разрешает неограниченное повторное использование, распространение и воспроизведение на любом носителе при условии, что оригинал работа правильно процитирована.Что такое секвенирование при долгом чтении?
Секвенирование ДНК — процесс считывания части или всей ДНК организма — помогает улучшить клиническую помощь в различных областях медицины, от редких заболеваний и рака до лечения инфекционных заболеваний.
Прогресс был ускорен развитием технологий высокопроизводительного секвенирования следующего поколения (NGS), которые способны считывать код миллионов небольших фрагментов ДНК параллельно. Это позволило ускорить секвенирование с увеличенной пропускной способностью при снижении затрат. В последние годы новые технологии, которые позволяют секвенировать более длинные цепи ДНК путем считывания отдельных молекул ДНК, получили развитие и стали более заметными.
В этом брифинге объясняется, что такое долгосрочное секвенирование (LRS) и чем оно отличается от установленного короткого секвенирования (SRS).Второй сопроводительный брифинг: «Долгосрочное секвенирование: готовы к работе в клинике?» описывает потенциал этих технологий для определения последовательности диагностики в клинических условиях и в этом контексте проблемы, связанные с внедрением технологии.
Самое необходимое
- Одиночные молекулы, «истинные» секвенсоры с длинным считыванием, позволяют производить считывания, которые значительно длиннее, чем те, которые возникают в результате SRS. У этого есть несколько неотъемлемых преимуществ
- LRS может секвенировать части генома, которые нельзя легко секвенировать с помощью секвенирования с коротким считыванием.Более длинные чтения с большей вероятностью будут выглядеть отчетливо по сравнению с более короткими чтениями, что позволяет их объединять вместе с меньшей двусмысленностью
- Двумя доминирующими производителями «настоящих» технологий долгого считывания являются Pacific Biosciences (PacBio) и Oxford Nanopore Technologies (Nanopore)
Что такое долгосрочное секвенирование?
Геном большинства организмов (включая человека) слишком длинный, чтобы его можно было секвенировать как одну непрерывную цепочку. С помощью секвенирования следующего поколения «коротких считываний» ДНК разбивается на короткие фрагменты, которые амплифицируются (копируются), а затем секвенируются для получения «считываний».Затем используются биоинформатические методы, чтобы соединить считанные данные, как головоломку, в непрерывную геномную последовательность.
ТехнологииLRS позволяет получать намного более длинные (> 10 000 п.о.) чтения секвенирования, чем широко используемые системы SRS (75-300 п.о.). Некоторые платформы для секвенирования с длинным считыванием (LRS) производили считывание последовательности 882 000 пар оснований 1 , при этом некоторые группы пользователей сообщали о считывании более 2 000 000 пар оснований (2 МБ) 2 ; однако более распространены длины чтения от 10 000 до 100 000 п.н.
True LRS — иногда их называют секвенаторами третьего поколения — непосредственно секвенируют отдельные молекулы ДНК в реальном времени, часто без необходимости амплификации. Такой подход прямого упорядочивания позволяет производить считывания, которые значительно длиннее, чем считывания, полученные в результате SRS. В других, «синтетических» подходах к секвенированию длинных считываний используется модифицированная обработка образцов и традиционный SRS для вычислительного восстановления длинных считываний из более коротких считываний секвенирования.True LRS представляет собой наибольший отход от широко используемых систем короткого чтения.
В настоящее время двумя доминирующими производителями «настоящих» технологий долгого считывания являются Pacific Biosciences (PacBio) и Oxford Nanopore Technologies (Nanopore). Обе компании разработали платформы для секвенирования нуклеиновых кислот (ДНК и РНК) «в реальном времени», которые быстрее, чем современные технологии короткого считывания.
Преимущества
Есть несколько неотъемлемых преимуществ использования более длинных чтений для исследования геномных данных; они могут иметь преимущества для клинического анализа генома.
- Сборка генома: Геном человека имеет длину более трех миллиардов пар оснований ДНК и содержит множество повторяющихся участков генетического кода. Подобно сложной головоломке, повторная сборка генома из коротких чтений может быть сложной задачей, поскольку многие фрагменты выглядят очень похожими без дополнительного контекста. Данные длительного чтения могут упростить эту задачу, поскольку операции чтения с большей вероятностью будут отличаться друг от друга, что позволяет объединить их вместе с меньшей неоднозначностью и ошибками. Улучшения в сборке генома помогают закрыть пробелы в наших знаниях о геноме и позволяют лучше понять генетические причины болезней.
- Обнаружение вариантов: некоторые особенности отдельных геномов особенно трудно обнаружить и количественно оценить с помощью технологий SRS, например: большие и сложные перестройки, большие вставки или делеции ДНК, повторяющиеся области, высокополиморфные области или области с низким нуклеотидным разнообразием ДНК. Длинные чтения могут охватывать более крупные части этих регионов, поэтому они могут обнаруживать больше этих вариантов, которые могут быть клинически значимыми. LRS может также усиливать «общегеномное» обнаружение определенных вариантов 3 .
- Фазирование гаплотипа: в таких областях, как репродуктивная медицина, может быть полезно знать, существуют ли генетические варианты на одной и той же копии хромосомы. Это можно определить с помощью процесса, известного как фазирование гаплотипа. Долговременные чтения могут предоставить информацию дальнего действия для определения гаплотипов без дополнительных статистических выводов, материнского / отцовского секвенирования или подготовки образца, что требуется для приблизительного определения фаз с использованием SRS.
Помимо выполнения длинных операций чтения, настоящие технологии LRS обладают и другими функциями, открывающими новые возможности.Среди них:
- Портативность: в отличие от других платформ для секвенирования, устройства Nanopore основаны на обнаружении электронных, а не оптических сигналов. Это позволяет им разрабатывать устройства размером с карту памяти (USB), что делает их очень портативными. Многие другие секвенсоры, включая подавляющее большинство систем SRS, представляют собой большие настольные или отдельно стоящие машины. Устройство Nanopore MinION использовалось для секвенирования проб в полевых условиях во время вспышек вирусов Эбола и Зика и даже использовалось в космосе.
- Секвенирование и скорость в реальном времени: по сравнению с фиксированным временем работы систем SRS и PacBio, и Oxford Nanopore предлагают более быстрые прогоны секвенирования. PacBio предоставляет возможности для быстрого секвенирования, которое может быть выполнено менее чем за 24 часа, от подготовки образца до анализа. Технологии нанопор позволяют проводить анализ в реальном времени и позволяют пользователю определять время выполнения эксперимента, давая пользователю возможность отслеживать сбор данных и начинать анализ по желанию. Это обеспечивает дополнительную гибкость и скорость, а также устраняет необходимость в пакетном секвенировании нескольких образцов, что в настоящее время требуется для экономичной SRS.Это особенно полезно при исследовании небольших геномов (например, многих патогенных микроорганизмов) или конкретных областей генома.
- Другие «омики»: для прямого секвенирования РНК использовались технологии длительного считывания. Они также могут позволить одновременное обнаружение эпигенетических модификаций (химических модификаций ДНК / РНК, влияющих на экспрессию генов), хотя требуется дополнительная биоинформатическая интерпретация. Для получения этой информации с использованием существующих систем SRS необходимо выполнить отдельные прогоны секвенирования.
Заключение
Неотъемлемые преимущества использования более длинных считываний для реконструкции и анализа генома, наряду с дополнительными потенциальными преимуществами настоящих систем LRS для анализа генома, могут быть полезны для диагностики нескольких заболеваний и нарушений. Однако системы LRS также создают свои проблемы и имеют некоторые ограничения; это и их потенциал для использования в клиническом секвенировании обсуждается в сопроводительном брифинге.
Благодарности
Мы благодарны д-ру Шехле Мохаммед за обзор этого брифинга и д-ру Саре Джеймс за исследование этой темы.
Заявление о конфликте интересов
PHG Foundation время от времени предоставляет аналитические услуги компании Oxford Nanopore Technologies (ONT). Однако этот брифинг является результатом независимого анализа и мнений PHG Foundation и не связан с какой-либо третьей стороной. Никакого внешнего финансирования для поддержки разработки этого брифинга получено не было, и ONT не принимал участия в его подготовке.
Список литературы
- Jain M. et al. Секвенирование нанопор и сборка генома человека со сверхдлинными считываниями. Nat Biotechnol, 2018.
- Payne A. et al. Наблюдение за китами с помощью BulkVis: графический просмотрщик файлов Oxford Nanopore для массовых файлов fast5. BioRxiv, 2018. https://doi.org/10.1101/312256
- Stancun MC. и другие. Картирование и фазирование структурных изменений в геномах пациентов с помощью секвенирования нанопор. Nat Comms, 2017.
Эмма Джонсон, Собия Раза
В издании Guardian’s Long Read, без жесткой формулы или географических ограничений
Composite, на котором представлены некоторые из любимых произведений Guardian в 2017 году: Братислав Миленкович; Дизайн Хранителя
Секс-роботы, насилие в Мосуле и план неминуемой смерти королевы Елизаветы.Это были одни из лучших сюжетов прошлого года на эклектичном длинном сайте The Guardian «Долгое чтение».
«У нас нет простой формулы», — говорит редактор по вводу в эксплуатацию Дэвид Вольф. «Но мы всегда ищем отличные истории, особенно те, которые происходят в мире идей».
Пристрастие The Long Read к статьям, освещающим дебаты, часто относящиеся к академическим кругам, отличает его от многих издателей нарративной журналистики. Например, любимые статьи редакторов в 2017 году включали аналитические статьи о неолиберализме, статистике и «эпохе подшучивания».«Но даже статьи, основанные на идеях, должны основываться на достоверных отчетах и повествовании», — говорит Вольф.
The Guardian всегда публиковала сюжеты вне новостей, но The Long Read была основана в 2014 году, чтобы обеспечить специальный дом для журнальных повествований. «По мере того, как статьи становились короче, а новости становились все быстрее, росла потребность в материалах, рассказывающих полную историю», — говорит Вольф.
Под управлением Джонатана Шайнина, бывшего редактора онлайн-новостей The New Yorker, лондонский сайт публикует три статьи в неделю, объемом от 4000 до 6000 слов.Около 20 процентов настроено в Великобритании. В остальном любая тема в любой точке мира — это честная игра. И если есть хоть какой-то секрет взлома, говорит Вольф, то он пишет хорошо: «Если мы получаем захватывающую подачу из первого предложения и чувствуем, что писатель имеет реальный контроль над материалом, это отменяет все».
Вольф делится другими советами по продвижению The Long Read ниже. Мы также внимательно рассмотрим подачу, которую он получил от Алекса Бласдела для профиля Тимоти Мортона a.к.а. «Философ-пророк антропоцена». Наш разговор отредактирован для большей ясности.
Есть ли в Великобритании более давние традиции по сравнению с США?
Американская традиция лонгформ довольно уникальна, но, безусловно, в Великобритании была история подобных изделий. Примером может служить Granta, редактированная Яном Джеком. Газета Independent также выпустила много замечательных авторов, которые впоследствии стали соавторами The New Yorker, таких как Ян Паркер и Зое Хеллер.Большая часть этой «секретной» давней традиции Великобритании либо не архивируется, либо закрывается платным доступом. Такие места, как London Review of Books, сегодня публикуют полные формы. Но это намного маргинальнее, чем в США
.Считаете ли вы The Long Read британским изданием?
Мы освещаем мир, но мы действительно хотим побудить людей думать о Великобритании как о богатой теме для длинных историй. Мы попытались описать британские учреждения таким образом, чтобы о них не обязательно писали раньше.Один из моих любимых был о пабе, который сопротивлялся разработчикам, пытающимся его закрыть; он содержал историю и философию британского паба. Но The Guardian — по-настоящему глобальное издание: обычно одна треть наших посетителей — британцы, одна треть — американцы и треть — остальной мир. Поэтому мы стараемся найти способы рассказывать истории, даже на британские темы, которые были бы интересны и доступны повсюду. Мы применяем то же правило к питчу из Техаса или Боливии.
Глядя на ваш обзор любимых историй за 2017 год, что выделялось в некоторых из них?
В пьесе о смерти королевы есть невероятное чувство тона и атмосферы.Проходит величественно, спокойно — язык идеально подобран для предмета. Этот жанр — описание того, чего не произошло, — одна из самых сложных вещей для реализации. В «Неблагодарном беженце» Дины Найери, который представляет собой смесь мемуаров и споров, написано впечатляюще, и есть сдержанная страсть, которая сразу же становится мощной. Статья Стивена Бурани о научных публикациях берет тему, которая звучит невероятно скучно, и превращает ее в удивительную бизнес-историю с невероятными персонажами.Если вам удастся убедить нас в том, что то, что сначала не кажется интересным, на самом деле является диким миром, это здорово для нас.
Есть ли части света или темы, о которых вы хотели бы видеть больше презентаций?
Мы редко получаем передачи из Латинской Америки. Удивительно, но мы получаем несколько презентаций из Европы, кроме Испании и России. В США, если в The New York Times появится какая-то отличная история, ее будут изучать три репортера.
«Если мы получаем убедительную презентацию из первого предложения и чувствуем, что писатель имеет реальный контроль над материалом, это отменяет все.
В Европе есть стандартные новости и политические репортажи, но из-за того, что нет культуры длинных форм, должно быть множество замечательных историй, которые не рассказывают. Одним из примеров является рассказ нашего корреспондента в Испании Джайлза Тремлетта о родителях, которые усыновили китаянку, а затем убили ее. Дело было невероятно известным в Испании, но в остальном неизвестно.Мы редко получаем презентации для деловых историй или профилей компаний. Мы хотели бы создать больше историй, подобных той, которую мы опубликовали в марте, о рынке люксовых колясок.
Как часто вы работаете с фрилансерами?
Сценаристы Guardian делают около одной трети статей, а остальные пишут фрилансеры.
Как часто вы присваиваете истории, основанные на холодных тонах?
Назначение историй, основанных на холодных тонах, относительно редко, отчасти потому, что они часто не соответствуют нашим потребностям. Два из них сработали, в результате появилась история о поддельном посольстве в Гане и одна о шеф-поваре, который был одним из пионеров молекулярной гастрономии и покончил с собой в 28 лет.Многие истории возникают из чатов с писателями, с которыми у нас есть отношения или с которыми мы связались после прочтения того, что они написали. Но если мы получаем многообещающую презентацию, мы часто остаемся на связи с писателем. Стоит отправить презентацию, даже если вы не уверены, что она правильная.
Какие типичные ошибки вы видите в презентациях?
Самая распространенная — это недостаточное знакомство с тем, что мы опубликовали. Простое упоминание пары вещей, которые мы сделали, сразу наводит меня на мысль, что этот человек задумался, почему эта история работает на нас.Мы также иногда получаем сообщения, в которых говорится: «Я из страны Икс, и вот что происходит. Хотите отчет об этом? » Лучшие презентации учитывают тот факт, что остальная часть газеты представляет собой новостные статьи и короткие статьи, а статья для длительного чтения должна быть отличной. Нам нужны большие истории, которые разворачиваются во времени, а не репортажи с места. Когда дело доходит до обсуждения тем, которые уже получили широкое освещение в СМИ, это всегда хороший знак, если автор может показать, чем эта конкретная история отличается от других.Писатели делают это на удивление редко, и это помогает выделиться на фоне других.
Что вы думаете об одновременной подаче заявок?
Я определенно в порядке. Тем не менее, иногда это может указывать на то, что вы недостаточно тщательно продумали, подходит ли он нам.
Сколько вы платите?
Он довольно сильно различается, но я могу сказать, что он конкурентоспособен.
Есть ли у вас бюджет на поездки?
Да, но мы сделаем только то, что требует длительных путешествий с писателем, с которым мы работали раньше или у которого есть опыт.
Каковы ваши предпочтения в отношении писателей, которые вам соответствуют?
У нас около 50 презентаций в неделю, а нас всего трое — я, Джонатан Шайнин и Клэр Лонгригг. Мы стараемся реагировать на каждую подачу, но я уверен, что есть моменты, которые мы упускаем. Если писатель не получал от нас известий в течение недели, он обязательно должен связаться с нами. Я всегда прошу людей писать по электронной почте, а не по телефону.
***
Мои вопросы — красным, ответы Волка — синим.Чтобы сначала прочитать презентацию без аннотаций, нажмите кнопку «Скрыть все аннотации», которую вы найдете под кнопками социальных сетей в правом верхнем меню.
Вы знали этого писателя, или это был холодный трюк? Он работал с Джонатаном Шайниным несколькими годами ранее в качестве редактора в Индии, поэтому мы знали его больше как редактора, чем писателя. Вы часто запускаете профили? Что делает хороший предмет? Нет, наверное, шесть-десять штук из 150 в год. Но мы рады получать презентации для профилей.Существует примерно два типа профилей: один — это человек, о котором вы уже слышали, например, наши профили президента Франции Эммануэля Макрона или ученого Ричарда Докинза. Другой — это человек, которого вы не обязательно знаете. Этот человек должен быть очаровательным и иметь интересную историю, но также должен освещать мир. Должна быть причина, выходящая за рамки необычных подробностей чьей-либо истории жизни. В этом случае центральная фигура была не только яркой и харизматичной, но и была способом проникновения в идею антропоцена.Философ-эколог Тимоти Мортон хочет, чтобы мы все «охладели». Было ли это открытием для вас эффективным способом привлечь ваше внимание? Вы не ожидаете, что предложение, которое начинается со слов «экологический философ», заканчивается словами «охренеть, холодок». Это предложение сразу предполагает, что писатель знает, что делает: он заинтересовал вас и удивил вас. Если бы он сразу начал с идеи антропоцена, это могло показаться утомительным. Но шок от первого предложения и размах второго дает вам отличный удар.Это не то, что вы могли бы ожидать от мыслителя, чьи все более влиятельные работы борются с катаклизмами — экстремальной погодой, затопленными городами, острой нехваткой ресурсов, ядерными осадками — которые возникли в антропоцене, новой геологической эпохе, в которой люди радикально меняют планету. . Вы слышали об этом философе? Я был знаком с его именем, но не читал его работ и толком не понимал, о чем он.
Но человек, который говорит, что наступление антропоцена означает осознание того, что «выхода нет» из «склепа», в котором мы живем, также находит в этом затруднительном положении карнавальное освобождение.«Вы думаете, что экологически чистая жизнь означает полную эффективность и чистоту», — недавно написал в Твиттере Мортон. «Неправильный. Это значит, что вы можете устроить дискотеку в любой комнате вашего дома ». Что вы думаете о включении этих цитат в начало? Это говорит о том, что автор уже проделал довольно много работы и думает о том, как сделать произведение интересным для того, кто еще не знаком с предметом. Они также дают вам представление о личности Мортона.
Этот твит был характерен для философии Мортона, которая исходила из самоочевидного, но затем резко отклонялась от проторенного пути.Его идиосинкразический образ мыслей, поглощающий все, от романтической поэзии и ультрасовременного саунд-арта до сверхглубоких скважин и «Бегущего по лезвию», теперь становится популярным. Мортон недавно был назван одним из пятидесяти самых влиятельных философов из ныне живущих. Его работа была подробно рассмотрена в американском выпуске Newsweek и процитирована в New York Times. Соруководитель Serpentine Ханс Ульрих Обрист недавно сказал читателям Vogue, что книги Мортона — самые важные произведения культуры.Повлияло ли на вас это внешнее подтверждение его важности? Насколько важен своевременный крючок? Писатель здесь явно осознает, что имя этого парня не нарицательное. Очевидно, что обзоры в Newsweek и комментарий Обриста помогают ответить на вопрос «почему этот парень». Мы работаем с довольно длительным сроком выполнения заказа, по крайней мере, месяц, но обычно от двух до четырех месяцев, мы обычно не ищем крючки как таковые. Мы хотим, чтобы все, что мы публикуем, было безотлагательным — это то, что важно сейчас, — без необходимости привязки к чему-то.Этот список способов, которыми он проникает в более массовую культуру, кажется хорошим способом сказать: «Этот человек актуален и важен». Но если кто-то появится в фильме, который выйдет на следующей неделе, это не сработает в наши сроки. Когда я говорил с Мортоном о возможности профилирования его, я спросил, какие песни будут в большой ротации на гипотетической дискотеке в его доме. Важно или необходимо заранее обезопасить доступ? Это зависит от обстоятельств, но я бы не сказал, что это важно.Если это ученый, который не подвергается постоянным нападкам, стоит посмотреть, можете ли вы позвонить им и запланировать возможности для отчета, но мы, конечно, не ожидаем, что вы этого сделаете. Если вы публикуете профиль известного человека или кого-то, кто находится в центре новостной статьи, это не обязательно, но стоит подумать, есть ли у вас шанс получить к нему доступ. В принципе, мы могли бы пойти на многое, но если вы хотите профилировать Бейонсе, вы должны показать нам, что вы, скорее всего, проведете с ней время с пользой.На вершине его сет-листа была «Hyperballad» — он определил микс «Subtle Abuse» — Бьорка, с которым он ведет долгую переписку и является соавтором. Хотя в настоящее время он пишет научную работу, в которой пытается объединить марксизм и темную экологию, у него также есть книга, выходящая в издании Penguin «Быть экологической», которая призвана очаровать широкую публику. Он сказал мне, что первое предложение звучит так: «Эта книга не содержит никаких экологических фактов». Почему было эффективно включить эти детали? Это точно скажет вам, что это за ботанический музыкант.Это меня успокаивает, что писатель получит характерный элемент профиля. Он должен уметь не только оживлять свои идеи, но и заставлять меня уходить с ощущением, будто я провел время с этим человеком и увидел мир его глазами. Это в стороне заставляет меня думать, что писатель хорошо замечает мелочи.
Во многом развитие мысли Мортона связано с его личной историей. Мортон родился в семье английской богемы и защитил докторскую диссертацию по романтической поэзии.В какой-то момент ему поставили диагноз депрессия. Попытки справиться с ситуацией открыли для Мортона целый ряд переживаний, от психоанализа до буддийской медитации, и его творчество стало более широким. Он сравнивает жизнь в депрессии с жизнью с трагическим знанием экологической катастрофы, которую мы создали: вы должны признать, «что вы работаете со сбоями, и этот сбой никуда не денется, потому что сбой на самом деле является частью того, как обстоят дела». После этого принятия, говорит Мортон, вы можете иногда переходить к чему-то, что больше похоже на игру.Почему было важно включить часть личной истории Мортона, и было ли это подходящим местом для этого фона? Конструктивно это наступает в хороший момент. Подача началась с того, что привлекло ваше внимание и дало представление об основных идеях этого парня и о том, почему он важен, а теперь в нем рассказывается его история. Писатель также хорошо связывает идеи этого парня с его личной жизнью. В лучших профилях чьи-то идеи и история жизни часто идут рука об руку, и связь между ними не кажется чрезмерно определенной или неуклюжей.
Позиция Мортона — что экологическая катастрофа, которую многие из нас опасаются, «уже произошла» и что процветание в эпоху антропоцена означает принятие ограничений нашей способности управлять миром — противопоставляется практически любому другому подходу к окружающей среде. Это не сделало его популярным среди других ученых. Некоторые профессиональные философы считают его в лучшем случае любопытством, а в худшем — мошенником. В одном из научных обзоров было указано на что-то «о продавце змеиного масла в прозе Мортона.Другой мыслитель утверждал, что Мортон и его небольшая когорта философских собратьев, которые являются активными блоггерами, преуспели лишь в создании «онлайн-оргии глупости». Другие утверждают, что он фатально оторван от экологических катастроф, нависших над глобальным югом: для белого человека средних лет с оксфордским образованием и университетским стажем — это хорошо и хорошо — защищать ветряные дискотеки и дружить с Бьорком — но как черт возьми, игра поможет фермерам в новых мисках для пыли в мире? Почему было важно включить голоса критиков? При продвижении профилей есть тенденция немного преувеличивать фигуру.Этот абзац сразу сигнализирует о том, что у писателя критический взгляд и он смотрит на этого парня отстраненно. Он знает о критике, и в этой статье он не назовет этого парня лучшим человеком с тех пор, как нарезал хлеб и проглотил всю его чушь. Я чувствую, что нахожусь в надежных руках. Это также приводит к конфликту: одни люди говорят, что этот парень блестящий мыслитель и освещает проблемы нашего времени, а другие говорят, что он идиот и мошенник — это звучит интересно.
В конечном счете, однако, я рассматриваю этот профиль как шанс описать мыслителя, идеи которого отражают и формируют то, как культура в целом думает о текущем состоянии мира, и рассказать историю идеи — антропоцена, — которая захватывает ненадежность, ужасность и странность того, что значит быть живым прямо сейчас.Что вы подумали об этом ближе? После нескольких абзацев о его жизненной истории и критике полезно вспомнить более широкую картину и идею антропоцена. Это напоминает мне, что речь идет не только об одном парне. Что произойдет после того, как вы получите такую сильную подачу? Обычно мы звоним или встречаемся с писателем и отправляем вопросы по логистике. Затем мы либо скажем «да», либо попросим автора набросать план рассказа так, как они его видят.
